
Mukowiscydoza to wrodzona, przewlekła choroba, w przebiegu której można dostrzec “efekt motyla” – uszkodzenie jednej, maleńkiej struktury prowadzi do kaskady objawów i powikłań, które są tak ciężkie, że skracają długość życia i znacznie obniżają jego jakość. O jakim defekcie mowa? Jakie objawy daje mukowiscydoza? Czy istnieją skuteczne metody diagnostyki i leczenia? Odpowiedzi znajdują się w poniższym artykule.
Spis treści:
- Mukowiscydoza – co to jest?
- Jakie są przyczyny mukowiscydozy?
- Mukowiscydoza – pierwsze objawy
- Mukowiscydoza – diagnostyka
- Mukowiscydoza – rokowania
- Objawy i leczenie mukowiscydozy – podsumowanie
Mukowiscydoza – co to jest?
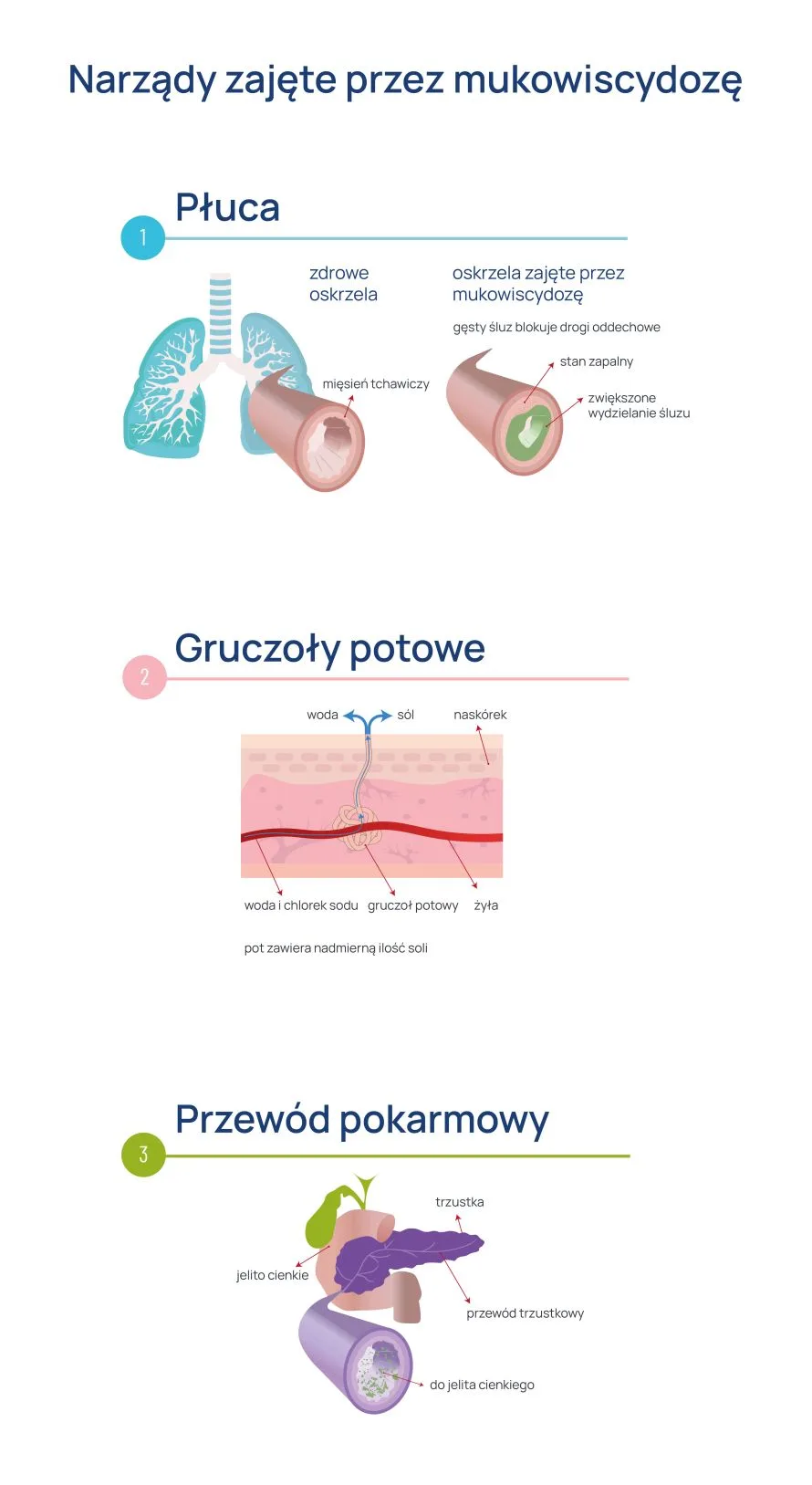
Mukowiscydoza to choroba jednogenowa, w której wskutek mutacji uszkodzeniu ulega gen kodujący białko błonowe CFTR. Białko to jest kanałem chlorkowym błony komórek nabłonkowych, regulatorem innych kanałów jonowych oraz odpowiada za transport wodorowęglanów.
Jego dysfunkcja sprawia, że gruczoły zewnątrzwydzielnicze (głównie w obrębie przewodu pokarmowego i układu oddechowego) produkują wydzielinę gęstą, lepką, uniemożliwiającą prawidłowe oczyszczanie śluzowo-rzęskowe, co niesie za sobą szereg poważnych konsekwencji.
Jakie są przyczyny mukowiscydozy?
Mukowiscydoza jest spowodowana patogennymi mutacjami w pojedynczym dużym genie zlokalizowanym na ludzkim chromosomie 7, który koduje białko transbłonowego regulatora przewodnictwa związanego z mukowiscydozą (CFTR).
Najczęstszą mutacją jest delecja trzech nukleotydów (“cegiełek” budujących geny) fenyloalaniny w miejscu 508 (około 70% pacjentów), jednak wyróżniono także wiele innych.
Mutacje genu CFTR można podzielić na sześć różnych klas, które z grubsza odpowiadają określonym typom dysfunkcji białka. Mutacje w klasach I–III powodują poważniejszą chorobę niż mutacje w klasach IV–VI. Jednak ich objawy kliniczne mogą się różnić, być może ze względu na działanie modyfikatorów genów.
W naszej szerokości geograficznej mukowiscydoza występuje z częstością około 1 na 3000 do 4000 żywych urodzeń, a około jedna na 25 do 30 osób jest nosicielem patogennej mutacji genu CFTR.
Mukowiscydoza – pierwsze objawy
Charakterystycznym, najwcześniejszym objawem jelitowym mukowiscydozy jest niedrożność smółkowa u noworodka, która powstaje w wyniku niedoboru enzymów trzustkowych.
Typowymi objawami mukowiscydozy u dzieci są brak prawidłowego oddania smółki, wzdęty brzuch i wymioty, występujące w pierwszych dobach życia dziecka. Objaw ten, bardzo charakterystyczny, jest jednak obecny jedynie u 5 do 20% chorych.
Na nieco późniejszych etapach życia objawem, który powinien zaniepokoić każdego rodzica i lekarza, jest opóźniony wzrost dziecka, pomimo prawidłowego apetytu i przyjmowania pokarmów.
Na mukowiscydozę u dzieci mogą wskazywać także nawracające zakażenia dolnych dróg oddechowych przebiegające z produkcją gęstej, trudnej do odkrztuszenia wydzieliny. Niejednokrotnie dochodzi do przewlekłego zapalenia zatok, bólów brzucha z towarzyszącym opóźnionym wzrostem i nieprawidłowym przybieraniem na masie.
Mukowiscydoza – objawy u dorosłych
Postępująca niewydolność narządów wewnętrznych prowadzi do szeregu poważnych konsekwencji w późniejszych latach życia. Wśród objawów mukowiscydozy u dorosłych istotnym problemem są częste, nawracające zakażenia dolnych dróg oddechowych.
Prowadzą one do stopniowej utraty funkcji płuc, co objawia się zwiększoną męczliwością, częstym odkrztuszaniem plwociny i tzw. palcami pałeczkowatymi. Przewlekła choroba płuc może z czasem prowadzić do powikłań kardiologicznych z przerostem mięśnia sercowego na czele. Z kolei powtarzające się epizody zapalenia zatok z czasem mogą skutkować rozwinięciem się polipów nosa.
Postępująca destrukcja trzustki prowadzi nie tylko do biegunki tłuszczowej, utraty masy ciała i bólów brzucha, ale także do cukrzycy – w trzustce bowiem znajdują się komórki produkujące insulinę.
Dysfunkcja wydzielnicza udziela się także w drogach żółciowych – u chorych na mukowiscydozę istotnie częściej dochodzi do kamicy żółciowej. Choroba ta może być przyczyną bezpłodności.
Mukowiscydoza – diagnostyka
W Polsce od 2009 roku istnieje program badań przesiewowych u noworodków, którego celem jest identyfikacja kilku częstych chorób wrodzonych, w tym mukowiscydozy. Diagnostyka składa się z kilku etapów. Po pobraniu kilku kropel krwi wysyła się próbkę do ośrodka specjalistycznego, w którym oznaczany jest tzw. immunoreaktywny trypsynogen.
W przypadku wyniku dodatniego pacjent jest informowany o konieczności poszerzenia diagnostyki. W dalszym etapie jest wykonywanie badanie genetyczne, a także test chlorków w pocie. W niektórych przypadkach wynik badania początkowego jest fałszywie ujemny, stąd diagnostyka będzie konieczna na późniejszych etapach życia.
Gdy są obecne objawy sugerujące mukowiscydozę, nie należy zwlekać z podzieleniem się wątpliwościami z pediatrą. Inną opcją jest wykonanie badania genetycznego – komercyjnie są dostępne testy badające od 160 do 170 najczęstszych mutacji w obrębie genu CFTR, jednak należy pamiętać, że i to badanie nie wyklucza w pełni mukowiscydozy.
W świetle obecnej wiedzy liczba znanych mutacji wynosi aż około 2000.

Mukowiscydoza – rokowania
Mukowiscydoza, jako wielonarządowa, ciężka choroba przewlekła, wymaga wielodyscyplinarnego podejścia. Terapia jest bardzo złożona, niemniej warto wspomnieć o wprowadzonej w kilku ostatnich latach grupie leków.
Chodzi o modyfikatory białka CFTR, które zrewolucjonizowały jakość życia i rokowanie pacjentów. W Polsce od 2020 roku są rozwijane programy lekowe w ramach ośrodków specjalistycznych. Obecnie są refundowane trzy preparaty o ściśle określonych kryteriach włączenia, co sprawia, że nie wszyscy pacjenci mogą być leczeni tą zaawansowaną grupą leków.
Szacuje się, że pacjenci z mukowiscydozą żyją do około czwartej dekady życia, zanim będą wymagali przeszczepienia płuc, które z kolei zapewnia średni czas przeżycia około 8,5 roku. Według najnowszych danych prawie 27% chorych nie przekroczy 30. roku życia, a prawie 50% wieku 40 lat.
Objawy i leczenie mukowiscydozy – podsumowanie
Mukowiscydoza jest chorobą wywołaną mutacją w genie CFTR, co sprawia, że organizm chorego wytwarza bardzo gęsty i lepki śluz w obrębie wielu narządów i tkanek, głównie płuc i przewodu pokarmowego.
Jest to przyczyną szeregu objawów i powikłań z postępującą utratą funkcji płuc na czele i w konsekwencji przedwczesnym zgonem. W wielu krajach, w tym w Polsce, istnieje program badań przesiewowych u wszystkich noworodków, co pozwala na wczesną diagnostykę i leczenie.
Nowoczesne terapie, wprowadzone w kilku ostatnich latach, według aktualnych danych istotnie poprawiają jakość życia i rokowanie, jednak nie wszyscy pacjenci kwalifikują się do tego typu postępowania.
Bibliografia
- Farrell PM, White TB, Ren CL, Hempstead SE, Accurso F, Derichs N, Howenstine M, McColley SA, Rock M, Rosenfeld M, Sermet-Gaudelus I, Southern KW, Marshall BC, Sosnay PR. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J Pediatr. 2017 Feb;181S:S4-S15.e1. doi: 10.1016/j.jpeds.2016.09.064. Erratum in: J Pediatr. 2017 May;184:243. PMID: 28129811.
- Naehrig S, Chao CM, Naehrlich L. Cystic Fibrosis. Dtsch Arztebl Int. 2017 Aug 21;114(33-34):564-574. doi: 10.3238/arztebl.2017.0564. PMID: 28855057; PMCID: PMC5596161.
- Konstan MW, Pasta DJ, VanDevanter DR, Wagener JS, Morgan WJ; Scientific Advisory Group and the Investigators and Coordinators of ESCF. Epidemiologic Study of Cystic Fibrosis: 25 years of observational research. Pediatr Pulmonol. 2021 May;56(5):823-836. doi: 10.1002/ppul.25248. Epub 2021 Jan 12. PMID: 33434406; PMCID: PMC9123916.
- Sanders DB, Fink AK. Background and Epidemiology. Pediatr Clin North Am. 2016 Aug;63(4):567-84. doi: 10.1016/j.pcl.2016.04.001. PMID: 27469176; PMCID: PMC4967225.
- Chen Q, Shen Y, Zheng J. A review of cystic fibrosis: Basic and clinical aspects. Animal Model Exp Med. 2021 Sep 16;4(3):220-232. doi: 10.1002/ame2.12180. PMID: 34557648; PMCID: PMC8446696.
- Link SL, Nayak RP. Review of Rapid Advances in Cystic Fibrosis. Mo Med. 2020 Nov-Dec;117(6):548-554. PMID: 33311787; PMCID: PMC7721430.
- Scotet V, L’Hostis C, Férec C. The Changing Epidemiology of Cystic Fibrosis: Incidence, Survival and Impact of the CFTR Gene Discovery. Genes (Basel). 2020 May 26;11(6):589. doi: 10.3390/genes11060589. PMID: 32466381; PMCID: PMC7348877.
- Regard L, Martin C, Burnet E, Da Silva J, Burgel PR. CFTR Modulators in People with Cystic Fibrosis: Real-World Evidence in France. Cells. 2022 May 28;11(11):1769. doi: 10.3390/cells11111769. PMID: 35681464; PMCID: PMC9179538.
- Durda-Masny M, Goździk-Spychalska J, John A, Czaiński W, Stróżewska W, Pawłowska N, Wlizło J, Batura-Gabryel H, Szwed A. The determinants of survival among adults with cystic fibrosis-a cohort study. J Physiol Anthropol. 2021 Nov 8;40(1):19. doi: 10.1186/s40101-021-00269-7. PMID: 34749804; PMCID: PMC8573904.
