
Spis treści
- Jak często występuje hipercholesterolemia rodzinna?
- Dlaczego w hipercholesterolemii rodzinnej obserwujemy podwyższone stężenie cholesterolu LDL?
- Jakie stężenie cholesterolu LDL powinno nasunąć podejrzenie hipercholesterolemii rodzinnej?
- Jak się wartości pozostałych parametrów profilu lipidowego w przypadku hipercholesterolemii rodzinnej?
- Jak leczy się hipercholesterolemię rodzinną?
Hipercholesterolemia rodzinna (ang. Familial hypercholesterolemia – FH) jest to zaburzenie lipidowe charakteryzujące się genetycznie uwarunkowanym, znaczenie podwyższonym stężeniem cholesterolu frakcji LDL (lipoproteiny o niskiej gęstości – ang. Low-Density Lipoprotein). Ponieważ podwyższone stężenie cholesterolu ma miejsce od urodzenia, bardzo często niezdiagnozowane prowadzi do wczesnego rozwoju miażdżycy oraz jej powikłań, takich jak zawał serca, udar mózgu czy nagłego zgonu sercowego dużo wcześniej niż w populacji zdrowej.
Jest to zaburzenie determinowane przez nieprawidłowy wariant jednego genu, dziedziczony w sposób mendlowski – w tym przypadku autosomalnie dominująco. Według zasad dziedziczenia mendlowego każdy człowiek posiada dwa warianty (allele) każdego genu dziedziczone po jednym od każdego ze swoich rodziców. Allele znajdują się na parach autosomów – chromosomów innych niż chromosomy płciowe X i Y. Jeśli dany allel jest dominujący, oznacza to, że cecha z nim powiązana ujawni się niezależnie czy w parze towarzyszy mu allel recesywny (taki człowiek jest tzw. heterozygotą) czy drugi dominujący (taki człowiek jest tzw. homozygotą recesywną). Praktyczną konsekwencją tego mechanizmu jest dużo częstsze występowanie heterozygot niż homozygot oraz większe nasilenie dziedziczonej cechy u homozygot.
Z tego względu istotny pod kątem klinicznym jest podział FH na:
- Heterozygotyczną hipercholesterolemię rodzinną (HeFH – Heterozygous Familial Hypercholesterolemia)
- Homozygotyczną hipercholesterolemię rodzinną (HoFH – Homozygous Familial Hypercholesterolemia)
Jak często występuje hipercholesterolemia rodzinna?
Heterozygotyczna hipercholesterolemia rodzinna jest stosunkowo częsta. Choć dawniej uważano, że występuje u 1 na 500 osób, obecnie dane wskazują na znacznie wyższą częstość – nawet 1 na 250 osób. Zakładając wielkość polskiej populacji z ostatniego spisu powszechnego GUS, w Polsce może żyć nawet 150 000 pacjentów z FH.
Homozygotyczna hipercholesterolemia rodzinna jest w porównaniu do heterozygotycznej bardzo rzadka, z szacowaną częstością występowania wynoszącą od 1 na 300 000 do 400 000 osób.
Dlaczego w hipercholesterolemii rodzinnej obserwujemy podwyższone stężenie cholesterolu LDL?
Za większość przypadków hipercholesterolemii rodzinnej odpowiadają nieprawidłowe warianty w obrębie trzech genów: LDLR (80-85% wszystkich przypadków FH), APOB (5-10% przypadków), PCSK9 (<1% przypadków).
LDLR (low density lipoprotein receptor) koduje receptor, który wiąże cząsteczki LDL na powierzchni komórek, po czym cząsteczki LDL są wchłaniane do komórki i rozkładane. Nieprawidłowe warianty w tym genie mogą prowadzić do braku lub obecności dysfunkcyjnego receptora LDL, co uniemożliwia efektywne usuwanie LDL z krwi, prowadząc do zwiększenia stężenia cholesterolu LDL.
APOB (apolipoproteina B) koduje apolipoproteinę B, która jest kluczowym składnikiem cząsteczek LDL umożliwiającym wiązanie z receptorem LDL na powierzchni komórki. Nieprawidłowe warianty APOB mogą zakłócić to wiązanie, co w podobny sposób znowu prowadzi do zwiększenia stężenia cholesterolu LDL w krwi.
Białka PCSK9 (konwertaza proproteiny subtylizyna/keksyna typu 9) kodowane przez gen oznaczony tym samymi skrótem, odpowiadaja za regulację degradacji receptora LDL. W tym wypadku dziedziczenia wariantu „nabycia funkcji” zwiększa się aktywność białka PCSK9, zmniejszając liczbę dostępnych receptorów dla LDL, powodując wzrost stężenia cholesterolu LDL.
Jakie stężenie cholesterolu LDL powinno nasunąć podejrzenie hipercholesterolemii rodzinnej?
Chociaż nie ma ujednoliconych międzynarodowo kryteriów diagnozy FH, opracowano kilka narzędzi diagnostycznych opierających się na kryteriach klinicznych i genetycznych, w których wielkość stężenia cholesterolu LDL odgrywa kluczową rolę.
W tabeli przedstawione kryteria rozpoznania FH według Dutch Lipid Clinic Network, obejmujące: wywiad rodzinny, charakterystyczne objawy w badaniu fizykalnym, stężenie LDL oraz potwierdzenie podłoża genetycznego.
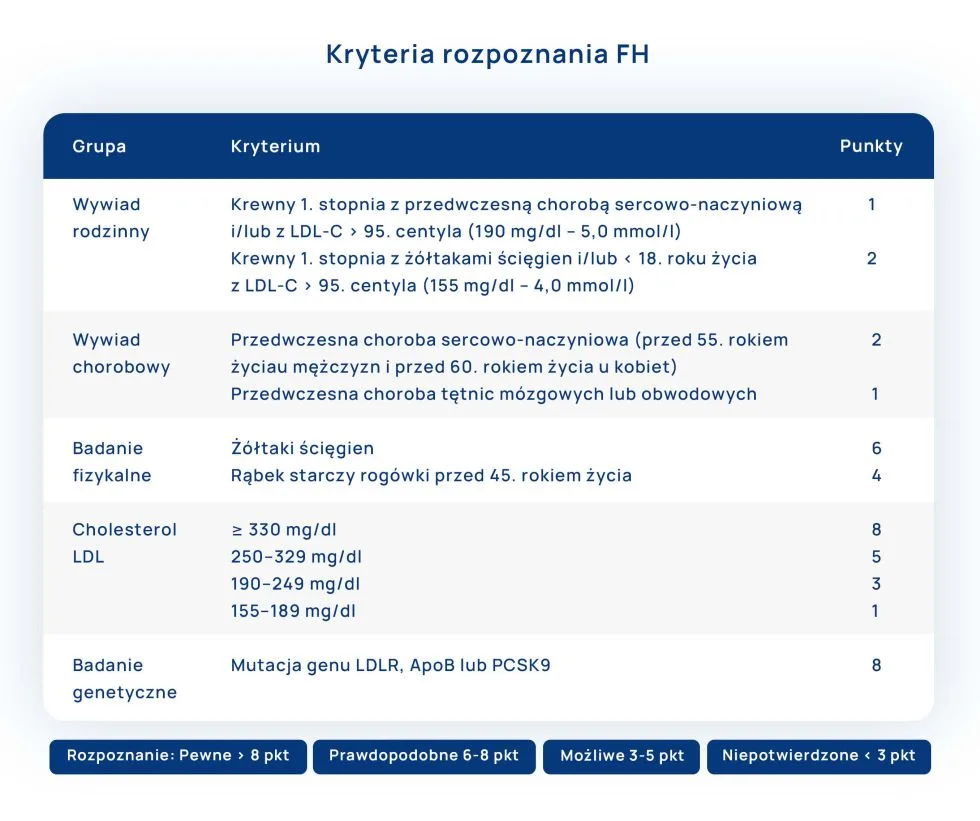
Jak można zaobserwować, po spełnieniu dodatkowych kryteriów pewną diagnozę hipercholesterolemię rodzinną można rozpoznać nawet w przypadku stężenia cholesterolu LDL ≥155 mg/dL. Odpowiednio prawdopodobieństwo odzwierciedlone punktacją rośnie przy wyższych stężeniach cholesterolu LDL. Zasadne jest rozważenie diagnozy heterozygotycznej postaci hipercholesterolemii rodzinnej u dzieci przy stwierdzeniu stężenia powyżej 160 mg/dL oraz u dorosłych, u których poziom cholesterolu LDL przekracza 190 mg/dL. Stężenie cholesterolu LDL ≥400 mg/dL może sugerować występowanie homozygotycznej hipercholesterolemii rodzinnej. Należy wspomnieć, że są to wartości dla pacjentów nieleczonych.

Bardzo często rozpoznanie hipercholesterolemii rodzinnej jest wyłącznie kliniczne, na podstawie wywiadu i wyników badań. Potwierdzenie genetyczne może być szczególnie przydatne w grupie młodych pacjentów, a także w diagnostyce członków rodziny. Brak stwierdzenia patologicznego wariantu w badaniu genetycznym nie wyklucza diagnozy FH, gdyż wariant występujący u pacjenta mógł nie być objęty badanym panelem lub w przypadku wielogenowego podłoża choroby.
Rozpoznanie hipercholesterolemii rodzinnej u członka rodziny powinno się wiązać z przesiewem kaskadowym polegającym na badaniu krewnych pierwszego stopnia (rodzice, rodzeństwo, dzieci). Jeśli wykryje się dotkniętego chorobą rodzica danej osoby, powinno się przebadać jak najwięcej krewnych z tej strony rodziny. Dzieci rodzeństwa dotkniętego rodzica powinny być również przebadane, ponieważ leczenie w dzieciństwie jest wskazane dla osób dotkniętych.

Jak się wartości pozostałych parametrów profilu lipidowego w przypadku hipercholesterolemii rodzinnej?
Podwyższenie stężenia cholesterolu całkowitego oraz nie-HDL będzie oczywistym skutkiem zwiększonego stężenia cholesterolu LDL. Typowe stężenie cholesterolu całkowitego u pacjentów z heterozygotyczną postacią może wynosić od 350 do 500 mg/dL. Wartości powyżej 500 mg/dL mogą sugerować postać homozygotyczną hipercholesterolemii rodzinnej.
Stężenia trójglicerydów oraz cholesterolu HDL (lipoproteiny o wysokiej gęstości, ang. High Density Lipoprotein) jest najczęściej w normie, a ich nieprawidłowe poziomy nie wykluczają diagnozy FH.
W przypadku oznaczenia stężenie apolipoproteiny B jej podwyższone stężenie będzie korelowało ze wzrostem stężenia cholesterolu LDL.
Coraz większą wagę przywiązuje się do oznaczenia stężenia lipoproteiny(a). Lipoproteina (Lp[a]) to kompleks składający się z lipoproteiny LDL oraz cząsteczki apolipoproteiny[a], które są ze sobą połączone na stałe. Liczba krążących cząsteczek lipoproteiny(a) jest w >90% determinowana genetycznie. Z uwagi na to, że przenosi ona również cholesterol, w przypadku zwiększonego stężenia tej cząsteczki może dawać to efekt podwyższonego stężenia cholesterolu LDL, dlatego ważne jest wzięcie pod uwagę Lp(a) przy ewentualnym wykluczeniu diagnozy FH.

Jak leczy się hipercholesterolemię rodzinną?
Głównym celem leczenia hipercholesterolemii rodzinnej jest zmniejszenie stężenia cholesterolu LDL w celu zmniejszenia ryzyka wystąpienia incydentu sercowo-naczyniowego.
Pacjenci z rozpoznaniem hipercholesterolemii rodzinnej bez dodatkowych czynników ryzyka oraz rozpoznania choroby sercowo-naczyniowej są traktowani jako pacjenci wyjściowo wysokiego ryzyka sercowo-naczyniowego, u których dąży się do uzyskania stężenia cholesterolu LDL <70 mg/dL oraz obniżenie wyjściowego stężenia o 50%. W przypadku rozwinięcia choroby sercowo naczyniowej pacjenci ci traktowani są jako grupa bardzo wysokiego ryzyka, u których dąży się do osiągnięcia stężenia cholesterolu LDL <55 mg/dL (przy obniżeniu wyjściowego stężenia o 50%)
Leczenie farmakologiczne obejmuje intensywną terapię lekami z grupy statyn, często w połączeniu z ezetymibem oraz inhibitorem PCSK9.
Niezależnie od postępowania farmakologicznego bardzo istotnym elementem leczenia jest modyfikacja diety oraz stylu życia.
W przypadku postaci homozygotycznej hipercholesterolemii rodzinnej często wymagane jest stosowanie połączenia terapii kilkoma grupami leków w maksymalnych tolerowanych dawkach, a wypadku nieosiągnięcia zakładanego efektu zastosowanie aferezy cząsteczek LDL – mechanicznego filtrowania osocza pacjenta z lipoprotein.
Piśmiennictwo
- Berberich AJ, Hegele RA. The complex molecular genetics of familial hypercholesterolaemia. Nature Reviews Cardiology. 2019;16(1):9-20.
- McGowan MP, Dehkordi SHH, Moriarty PM, Duell PB. Diagnosis and Treatment of Heterozygous Familial Hypercholesterolemia. Journal of the American Heart Association. 2019;8(24):e013225.
- Visseren FLJ, Mach F, Smulders YM, et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice: Developed by the Task Force for cardiovascular disease prevention in clinical practice with representatives of the European Society of Cardiology and 12 medical societies With the special contribution of the European Association of Preventive Cardiology (EAPC). European Heart Journal. 2021;42(34):3227-3337.
- Banach M, Burchardt P, Chlebus K, et al. Wytyczne PTL/KLRWP/PTK/PTDL/PTD/PTNT diagnostyki i leczenia zaburzeń lipidowych w Polsce 2021. Nadciśnienie Tętnicze w Praktyce. 2021;7(3):113-122.
