
Spis treści
- Rodzaje choroby Alzheimera
- Stadia choroby Alzheimera
- Diagnostyka choroby Alzheimera – badania obrazowe i laboratoryjne
- Genetyka choroby Alzheimera
Otępienie to stan chorobowy, w którym dochodzi do upośledzenia funkcji intelektualnych objawiających się problemami z pamięcią, spowolnieniem myślenia, czy też zaburzeniami zachowania. Do grupy chorób otępiennych zalicza się chorobę Alzheimera, otępienie czołowo-skroniowe, otępienie z ciałami Lewy’ego, oraz otępienie naczyniopochodne. Mechanizm powstawania trzech pierwszych związany jest z patologicznym gromadzeniem się w strukturach mózgu określonych białek, natomiast otępienie naczyniopochodne związane jest z niedokrwieniem mózgu w następstwie patologicznych zmian w naczyniach mózgowych takich jak przebyty udar mózgu, miażdżyca naczyń mózgowych czy nadciśnienie tętnicze.
Najczęstszą chorobą otępienną jest choroba Alzheimera (ang. Alzheimer’s disease, AD) powodująca poważne deficyty poznawcze i znacznie utrudniająca codzienne funkcjonowanie. W grupie pacjentów powyżej 65. roku życia jest ona najczęściej rozpoznawanym zespołem otępiennym, a częstość jej występowania rośnie z wiekiem.
Rodzaje choroby Alzheimera
Choroba Alzheimera (AD) może mieć postać sporadyczną (sporadic Alzheimer’s disease, SAD) lub rodzinną (family Alzheimer’s disease, FAD), gdy u co najmniej dwóch członków rodziny wcześniej rozpoznano chorobę Alzheimera. Natomiast w zależności od wieku wystąpienia pierwszych objawów u chorego wyróżnia się postać wczesną (early-onset Alzheimer’s disease, EOAD, pierwsze objawy przed 65. rokiem życia) lub późną (late-onset Alzheimer’s disease, LOAD, objawy po 65. roku życia). Postać rodzinna choroby Alzheimera jest rzadsza, dotyczy według różnych doniesień literaturowych około 15-40% wszystkich chorych. Zdecydowaną większość stanowi forma sporadyczna o wieloczynnikowej etiologii i złożonym sposobie dziedziczenia.
Stadia choroby Alzheimera
W chorobie Alzheimera wyróżnia się trzy stadia choroby:
- Stadium pierwsze związane z postępującymi problemami z pamięcią skutkujące wycofaniem się chorego z życia społecznego;
- Stadium drugie, gdzie do wyżej wymienionych zaburzeń dołączają również urojenia czy halucynacje;
- Stadium trzecie, w którym chory ze względu na duże deficyty funkcji poznawczych nie jest zdolny do samodzielnej egzystencji.

Diagnostyka choroby Alzheimera – badania obrazowe i laboratoryjne
U pacjentów, u których podejrzewa się chorobę Alzheimera, należy wykonać szereg badań diagnostycznych. Należą do nich badania laboratoryjne, obrazowe, psychologiczne (ocena stanu psychicznego wg. skali MMSE, Mini Mental State Examination) oraz neurologiczne. Ze względu na fakt, iż wiele nieprawidłowości w wynikach badań biochemicznych może powodować zaburzenia poznawcze i wpływać na pojawienie się otępienia bardzo ważną rolę w diagnostyce choroby Alzheimera spełniają podstawowe badania laboratoryjne, ze szczególnym uwzględnieniem morfologii krwi obwodowej, badań biochemicznych (poziomu hormonów tarczycy, lipidogramu, stężenia kreatyniny, mocznika, witaminy B12, kwasu foliowego, żelaza, sodu, potasu, wapnia, chlorków oraz enzymów wątrobowych). Wskazane jest również wykonanie ogólnego badania moczu oraz badań serologicznych w celu wykluczenia zakażeń drobnoustrojami patogennymi.
Uzupełnieniem badań laboratoryjnych jest diagnostyka obrazowa struktur mózgu tj. pozytonowa emisyjna tomografia komputerowa (PET), rezonans magnetyczny (MRI) oraz funkcjonalny rezonans magnetyczny (fMRI), dzięki którym można ocenić struktury mózgowia i oszacować czy nie doszło do odkładania się w nim patologicznych złogów będących biomarkerami choroby Alzheimera. Jeśli badanie obrazowe ujawnia nieprawidłowości, poszerza się diagnostykę o badania laboratoryjne płynu mózgowo-rdzeniowego. Poszukuje się wtedy dwóch form białek: amyloidu β oraz białka tau.
Pierwsze z nich posiada dwie izoformy: rozpuszczalną (Amyloid β40) oraz nierozpuszczalną (Amyloid β42). To właśnie patologiczna agregacja nierozpuszczalnej formy amyloidu β w strukturach mózgu, w postaci charakterystycznych blaszek amyloidowych upośledza funkcje neuronów, prowadząc do nieprawidłowych procesów oczyszczania przestrzeni międzykomórkowych przez komórki mikrogleju. Konsekwencją gromadzenia się blaszek amyloidu są zaburzenia w funkcjonowaniu mózgu prowadzące do powstania zaburzeń poznawczych. Białko tau występuje w komórkach nerwowych i bierze udział w formowaniu cytoszkieletu, wspomagającego również transport substancji odżywczych wzdłuż komórek nerwowych (transport aksonalny). Nagromadzenie dużych ilości białka, w wyniku jego nadmiernej fosforylacji prowadzi do tworzenia tzw. splotów neurofibrylarnych, czyli poskręcanych fragmentów białka w komórkach nerwowych, co zaburza ich zdolność do przekazywania sygnałów.
Połączenie wszystkich powyższych badań stanowi złoty standard w diagnostyce choroby Alzheimera i pozwala na postawienie rozpoznania i monitorowanie choroby.
Test zegara

Genetyka choroby Alzheimera
Badania biochemiczne w chorobie Alzheimera i odkrycie patologicznych form białka amyloidu β oraz białka tau odpowiadających za pojawienie się objawów tego schorzenia stały się punktem zwrotnym w poszukiwaniach genetycznego podłoża tej choroby. Zapoczątkowało to badania molekularne nad analizą sekwencji genów odpowiedzialnych za ich produkcję w organizmie. Pierwsze badania genetyczne w chorobie Alzheimera dotyczyły 4 genów: PSEN1, PSEN2, APP oraz APOE.

Gen PSEN1 oraz PSEN2 koduje białka preseniliny, które wchodzą w skład dużego kompleksu białkowego odpowiedzialnego za aktywność γ-sekretazy i biorą udział w przekazywaniu sygnałów molekularnych w różnych systemach komórkowych. Patogenne warianty sekwencji genów PSEN1 i PSEN2 zaburzają proces prawidłowego dojrzewania cząsteczki prekursora amyloidu β, a co za tym idzie do nadmiernego wytwarzania patologicznej, nierozpuszczalnej formy amyloidu β42. Dzięki wielu dużym badaniom na pacjentach z rodzinną postacią choroby Alzheimera oszacowano, że najczęstsze miejsca występowania mutacji punktowych w genie PSEN1 obejmują eksony od 5 do 12, a częstość ich występowania waha się pomiędzy 18 a 50%. Natomiast mutacje w genie PSEN2 obejmują eksony od 3 do 11, występują zdecydowanie rzadziej i związane są obok choroby Alzheimera z innymi postaciami chorób otępiennych. Mutacje w genach presenilin cechuje 100% penetracja.

Gen APP koduje białko będące prekursorem amyloidu β. Pierwszą mutację patogenną w genie APP wykryto w 1990 r. Wszystkie zidentyfikowane mutacje w genie APP występowały w pobliżu miejsc rozpoznawanych przez α-, β-, γ- sekretazy, udowodniono w ten sposób, że mają one kluczowe znaczenie w procesie dojrzewania produkowanego białka prekursora amyloidu i tworzenie się amyloidu β. Większość z mutacji zaburza aktywność sekretaz, prowadząc do nadmiernego wytwarzania toksycznego dla neuronów amyloidu β42, który odkłada się w postaci neurotoksycznych blaszek w mózgu pacjenta.

Gen APOE, odpowiedzialny za metabolizm lipidów, w tym cholesterolu, występuje w trzech głównych izoformach: e2, e3, e4, różniących się między sobą położeniem aminokwasów argininy i cysteiny w pozycjach: 112 i 158. W zależności od obserwowanego genotypu (Tabela 1) obserwujemy różne spektrum objawów u pacjentów. Przy czym występowanie izoformy e4 genu APOE jest głównym genetycznym czynnikiem ryzyka wystąpienia późnej formy choroby Alzheimera (LOAD), zwiększającym ryzyko 3-krotnie u nosicieli tej formy w postaci heterozygotycznych i 15-krotnie u nosicieli homozygotycznych. Uznano, więc że APOE może stanowić marker predykcyjny w przypadku prognozowania wystąpienia choroby Alzheimera, ale nie powinien być jedynym analizowanym parametrem, ze względu na obecność APOE e4/e4 również u zdrowych osób, u których nie rozwinęło się otępienie.

W ostatnich latach badania prowadzone na kilku dużych populacjach chorych potwierdziły kilka zależności pomiędzy 4 genami o udowodnionym udziale w powstaniu i rozwoju choroby Alzheimera. Wiadomo, iż status alleli genu APOE nie wpływa na wiek początku choroby w przypadkach wczesnej postaci choroby Alzheimera z obecnością patogennego wariantu sekwencji DNA w genie PSEN1, ma natomiast znaczenie u pacjentów obecnością patogennego wariantu sekwencji DNA w genie APP. Mutacje PSEN1 prowadzą do nadmiernego wytwarzania amyloidu β42, co zostało uznane w ostatnich latach za jeden z czynników patogennych w rodzinnej postaci choroby Alzheimera.
Pomimo tych doniesień wiele mechanizmów prowadzących do występowania choroby Alzheimera pozostaje niejasnych. Nowe techniki badawcze, zwłaszcza sekwencjonowanie następnej generacji (NGS, Next Generation Sequencing) pozwalają obecnie na badanie dużych grup chorych i wykonywania badań panelowych wielu genów jednocześnie. Dzięki temu liczba genów o udowodnionym wpływie na patogenezę choroby Alzheimera i innych zespołów otępiennych cały czas się powiększa. W opublikowanym w czasopiśmie Nature w 2022 roku artykule autorzy wytypowali około 75 nowych genów mających związek z zespołami otępiennymi, w tym z chorobą Alzheimera. Wśród nich obok znanych wcześniej genów APP, PSEN1, PSEN2 i APOE znalazły m.in.: SORT1, ADAM17, PRKD3, NCK2, WDR12, MME, IDUA, RHOH, ANKH, COX7C, TNIP1, RASGEF1C, HS3ST5, UMAD1, ICA1, TMEM106B, JAZF1, SEC61G, CTSB, SHARPIN, ABCA1, ANK3, TSPAN14, BLNK, SNX1, CTSH, DOC2A, MAF, FOXF1, PRDM7, WDR81, MYO15A, SIGLEC11, LILRB2, RBCK1, SLC2A4RG, IGH gene cluster, TREM2, INPP5D, CD2AP, PTK2B, CLU, NOTCH3 PLCG2 czy ABI3. Badania te pozwoliły też określić sposób dziedziczenia niektórych patogennych wariantów. I tak w przypadkach rodzinnej formy choroby Alzheimera przeważał dominujący sposób dziedziczenia, natomiast w formach sporadyczny choroby Alzheimera rodzaj dziedziczenia recesywny lub patogenne warianty pojawiały się do novo.
W tabeli poniżej przedstawiono niektóre geny zaangażowane w mechanizmy powstawania choroby Alzheimera wraz z lokalizacją na chromosomach człowieka i sposobem dziedziczenia.
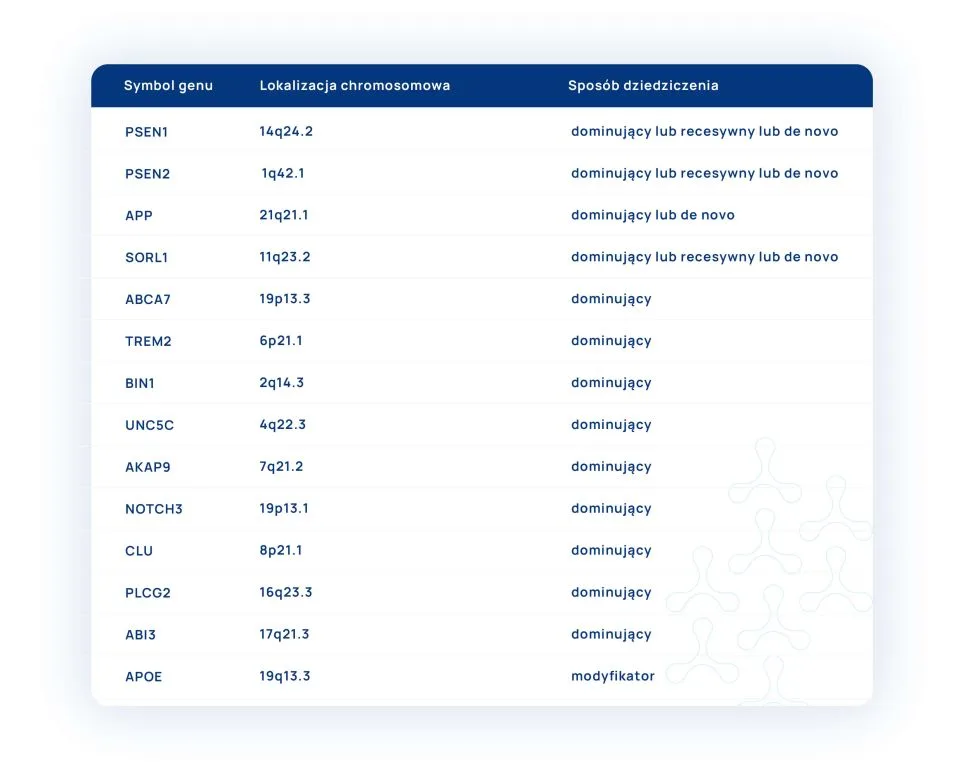
Reasumując, prowadzone wieloośrodkowe badania z zastosowaniem nowoczesnych technik badawczych dostarczyły w ostatnim dziesięcioleciu wielu nowych informacji o podłożu genetycznym i sposobie dziedziczenia choroby Alzheimera otwierając jednocześnie nowe możliwości diagnostyczne pacjentom i ich rodzinom. Ze względu na fakt, że choroba ta dotyka dużej części społeczeństwa wczesne wykrycie choroby i wdrożenie skutecznej celowanej terapii daje szansę chorym na skuteczną terapię i zapobiega wykluczeniu z życia społecznego.
Piśmiennictwo:
- Pfeffer A. Choroba Alzheimera – obraz kliniczny, rozpoznawanie, możliwości terapeutyczne zaburzeń poznawczych. Przewodnik Lekarza/Guide for GPs. 2004:70-78.
- Kowalska A. Genetyka zespołów otępiennych. Część 3: podłoże molekularne wieloczynnikowego dziedziczenia postaci sporadycznej choroby Alzheimera. Postepy Hig Med Dosw 2009; 63 : 577-582
- Prusiński A., Neurologia praktyczna, PZWL, Warszawa 2001
- Wieczorowska-TobisK., Talarska D., Geriatria i pielęgniarstwo geriatryczne podręcznik dla studiów medycznych, PZWL, Warszawa 2008
- Barcikowska-Kotowicz M. Obraz kliniczny choroby Alzheimera – charakterystyka trzech stadiów choroby. W: Sytuacja osób chorych na chorobę Alzheimera. Raport RPO. Szczudlik A (red.). Polskie Towarzystwo Alzheimerowskie, Warszawa 2014; 19-21.
- Wallin A.: Current definition and classification of dementia diseases. Acta Neurol. Scand. Suppl., 1996; 168: 39-44
- Bugaj A, Jermakow N. Mechanisms underlying Alzheimer’s disease. Neuropsychiatria i Neuropsychologia/Neuropsychiatry and Neuropsychology. 2016;11(3):85-92. doi:10.5114/nan.2016.63650.
- Mandecka M, Budziszewska M, Barczak A, Pepłońska B, Chodakowska-Żebrowska M, Filipek-Gliszczyńska A, Nesteruk M, Styczyńska M, Barcikowska M, Gabryelewicz T. Association between Cerebrospinal Fluid Biomarkers for Alzheimer’sDisease: APOE Genotypes and AuditoryVerbal Learning Task in Subjective Cognitive Decline, Mild Cognitive Impairment, and Alzheimer’s Disease JAD 2016, 54, 157 -168.
- Hoogmartens, J, Cacace, R, Van Broeckhoven, C. Insight into the genetic etiology of Alzheimer’s disease: A comprehensive review of the role of rare variants. Alzheimer’s Dement. 2021; 13:e12155.
- Roses A.D., Strittmatter W.J., Pericak-Vance M.A., Corder E.H., Saunders A.M., Schmechel D.E.: Clinical application of apolipoprotein E genotyping to Alzheimer’s disease. Lancet, 1994; 343: 1564-1565
- Selkoe D.J., Podlisny M.B.: Deciphering the genetic basis of Alzheimer’s disease. Annu. Rev. Genomics Hum. Genet., 2002; 3: 67–99
- Wehr H., Parnowski T., Puzyński S, Bednarska-Makaruk M., Bisko M., Kotapka-Minc S., Rodo M., Wołkowska M.: Apolipoprotein E genotype and lipid and lipoprotein levels in dementia. Dement. Geriatr. Cogn. Disord., 2000; 11: 70-73
- Sawamura N., Morishima-Kawashima M., Waki H., Kobayashi K., Kuramochi T., Frosch M.P., Ding K., Ito M., Kim T.W., Tanzi R.E., Oyama F., Tabira T., Ando S., Ihara Y.: Mutant presenilin 2 transgenic mice. A large increase in the levels of Aβ 42 is presumably associated with the low density membrane domain that contains decreased levels of glycerophospholipids and sphingomyelin. J. Biol. Chem., 2000; 275: 27901-27908
- Bellenguez, C., Küçükali, F., Jansen, I.E. et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet 54, 412–436 (2022).
