Czynniki ryzyka nieprawidłowej pracy tarczycy u kobiet w wieku prokreacyjnym, ich wpływ na możliwości zajścia w ciążę i donoszenia zdrowego dziecka.
Spis treści
- Znaczenie występowania przeciwciał przeciwtarczycowych przed ciążą
- Wartości referencyjne tyreotropiny (TSH) u kobiet w okresie przedkoncepcyjnym
- Czynniki ryzyka dysfunkcji tarczycy
Prawidłowe funkcjonowanie tarczycy jest nieodzownym warunkiem zajścia w ciążę oraz donoszenia zdrowego dziecka. Badania wskazują, że kobiety z autoimmunologicznym zapaleniem tarczycy częściej doświadczają problemów związanych z niepłodnością, poronieniami i porodami przedwczesnymi. Nieprawidłowości w układzie immunologicznym matki oddziałują na płód, osłabiają tolerancję immunologiczną pomiędzy matką i płodem, niekorzystnie rzutują na przebieg ciąży.
Dlatego każda kobieta w wieku prokreacyjnym, która planuje zajście w ciążę, powinna mieć pewność, że jej tarczyca pracuje prawidłowo i że nie występują u niej czynniki ryzyka dysfunkcji tego narządu. Artykuł omawia ścieżkę diagnostyczną dysfunkcji tarczycy u kobiet planujących ciążę i leczonych z powodu niepłodności, oraz niektóre parametry o znaczeniu predykcyjnym dla wystąpienia nieprawidłowości tarczycy (obniżone stężenie TSH, występowanie przeciwciał tarczycowych, itp.).
Znaczenie występowania przeciwciał przeciwtarczycowych przed ciążą
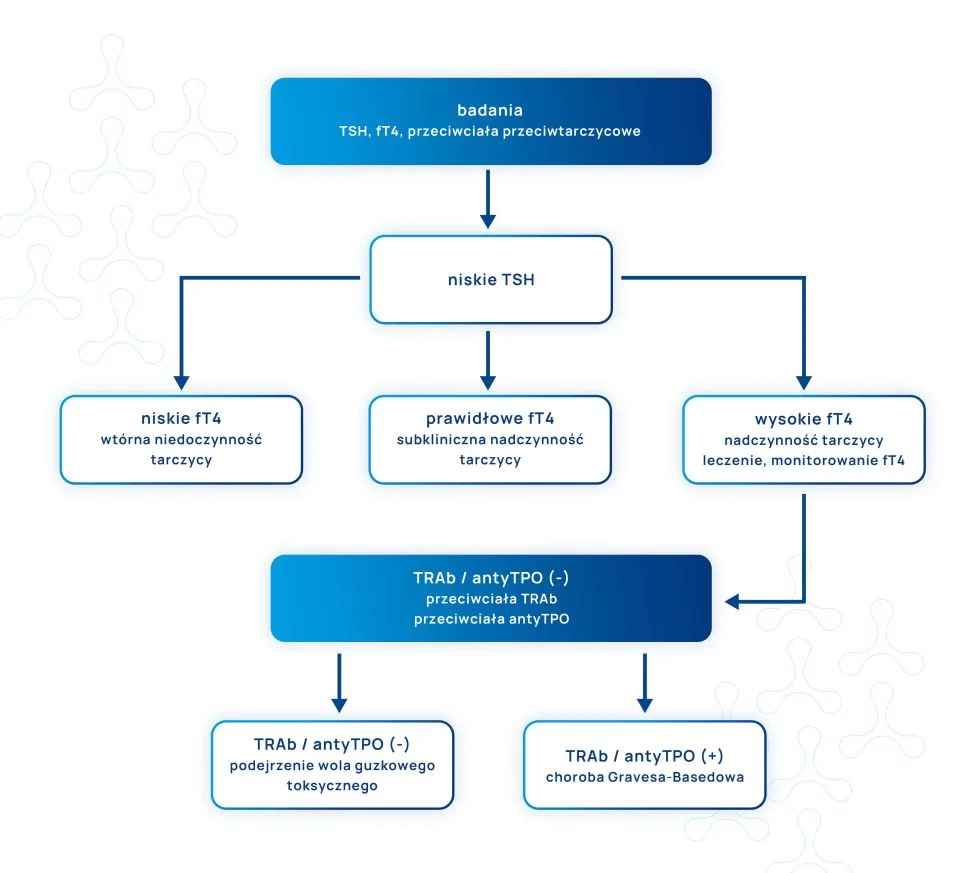
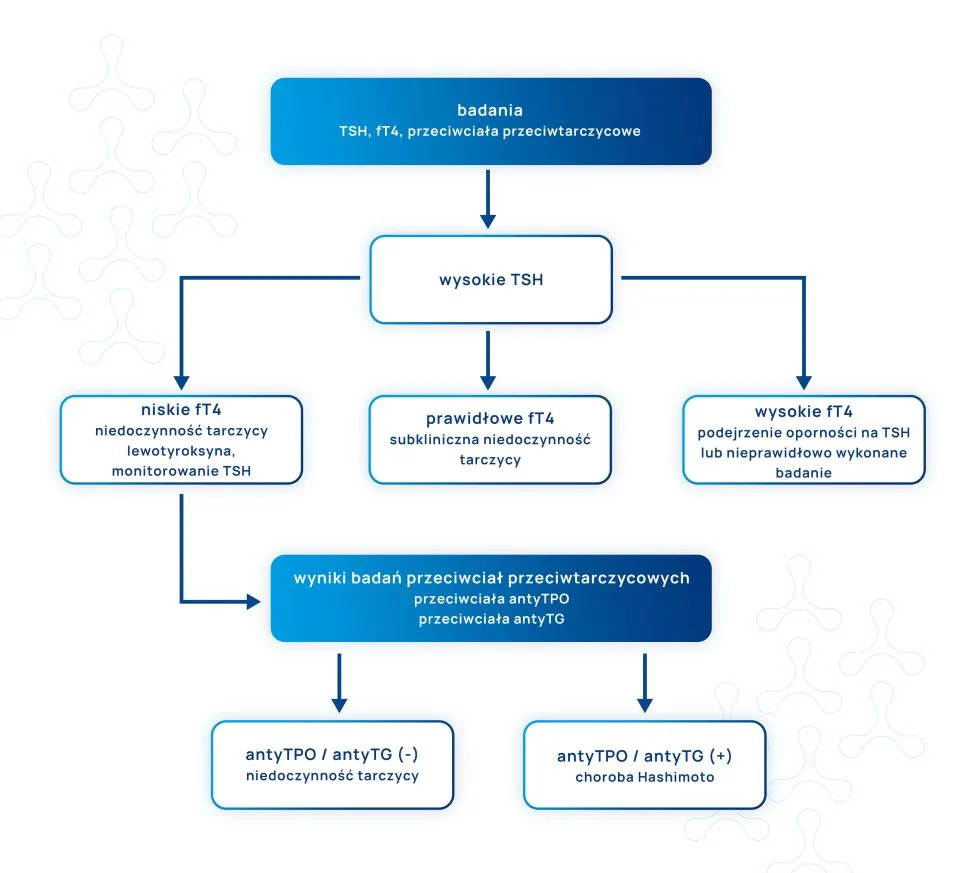
Przeciwciała przeciwtarczycowe – antyTPO (przeciw peroksydazie tarczycowej) i antyTG (przeciw tyreoglobulinie) – występują u ok. 20% kobiet w wieku prokreacyjnym i u ok. 15% kobiet ciężarnych. Należy podkreślić, że ich obecność jest silnie związana z występowaniem dysfunkcji tarczycy lub – jeśli w danej chwili funkcja tarczycy jest prawidłowa – jest to czynnik prognostyczny wystąpienia zaburzeń jej pracy.
Niestety, ponieważ 5-10% kobiet z dodatnimi przeciwciałami przeciwtarczycowymi nie wykazuje zaburzeń czynności tarczycy, w codziennej praktyce nie są one identyfikowane. Badania wykazują, iż obecność przeciwciał przeciwtarczycowych u kobiet w wieku rozrodczym łączy się z dużym prawdopodobieństwem powstania zaburzeń pracy tarczycy, szczególnie w postaci niedoczynności, co może być zagrożeniem dla donoszenia ciąży. Dlatego nieprawidłowa czynność tarczycy u kobiet w wieku prokreacyjnym oraz u kobiet w ciąży powinna być powodem do niepokoju i taki stan powinien być jak najszybciej identyfikowany.
W jednym z badań przeprowadzonych w Danii wzięło udział 825 kobiet z niewyjaśnioną, nawracającą utratą ciąży w wywiadzie. Okazało się, iż czynnikiem, który wywierał niekorzystny wpływ na powodzenie ciąży i urodzenie żywego dziecka był dodatni wynik testu antyTPO. Prawidłowa diagnoza i leczenie niedoczynności tarczycy hormonem T4 zwiększało szansę na żywy poród.
Badanie wspiera tezę, iż u kobiet przed ciążą i w ciąży, powinny być przeprowadzanie badania przesiewowe w kierunku wykrywania przeciwciał przeciwtarczycowych antyTPO i antyTG. W przypadku ich wykrycia następnym krokiem powinno być:
- oznaczanie poziomu TSH co 6 miesięcy – u kobiet planujących ciążę z podwyższonymi przeciwciałami antyTPO i/lub antyTG, ale z prawidłową czynnością tarczycy (eutyrozą);
- oznaczenie poziomu TSH w momencie stwierdzenia ciąży u kobiet z podwyższonymi przeciwciałami antyTPO i/lub antyTG
- oznaczanie poziomu TSH co 4 tygodnie u kobiet w pierwszej połowie ciąży, gdy mają podwyższone stężenie przeciwciał antyTPO i/lub antyTG.

Wartości referencyjne tyreotropiny (TSH) u kobiet w okresie przedkoncepcyjnym
Aktualne wytyczne dotyczące wartości referencyjnych TSH (hormon tyreotropowy, tyreotropina) rekomendują utrzymanie tego hormonu na poziomie < 2,5 mIU/L w okresie przedkoncepcyjnym oraz u kobiet w ciąży. Widełki dla ogółu populacji są szersze, górna granica TSH określana jest na ok. 4,0-4,5 (w zależności od laboratorium). Badania jednak jasno pokazują, iż wartości do 2,5 są optymalne, jeśli kobieta ma donosić zdrową ciążę. Niestety te wytyczne nie dotyczą ogółu kobiet w wieku rozrodczym, ok. 25% z nich ma TSH ≥ 2,5 mIU/L, co jest określane jako tzw. wartości wysokie, prawidłowe. Wiąże się to ze zwiększoną częstością poronień, przedwczesnych porodów lub innych nieprawidłowości metabolicznych, które mogą przyczynić się do niepowodzenia ciąży.
Dlatego z punktu widzenia możliwości zajścia w ciążę, a następnie dobrostanu i prawidłowego rozwoju dziecka wszystkie kobiety w okresie reprodukcyjnym powinny mieć TSH ≤ 2,5 mIU/L.
Dodatkowo należy pamiętać, że oznaczanie poziomu TSH powinno być badaniem rutynownym u wszystkich pacjentek diagnozowanych z powodu niepłodności.
Czynniki ryzyka dysfunkcji tarczycy:
a) niemodyfikowalne – niemożliwe do wyeliminowania,
b) modyfikowalne – związane głównie ze stylem życia, możliwe do skorygowania.
Utrzymywanie tarczycy w optymalnej formie zależy od wielu czynników, na część z nich każda pacjentka przed ciążą i w ciąży ma wpływ i może je korygować.
Do modyfikowalnych czynników ryzyka dysfunkcji tarczycy należą:
- niedobór jodu – kobiety w ciąży powinny suplementować jod, jednak warto zadbać o jego poziom już przed ciążą i monitorować stężenie jodu w organizmie. Badaniem, które najlepiej odzwierciedla stężenie jodu jest oznaczanie jego poziomu w moczu;
- niedobór żelaza – niedokrwistość z niedoboru żelaza i zaburzenia gospodarki żelazowej są powszechne w ciąży, ale u wielu kobiet występują już przed ciążą. Monitorowanie poziomu żelaza i parametrów gospodarki żelazem (ferrytyna) należą do badań rutynowych w ciąży, ale powinny na nie zwrócić uwagę wszystkie kobiety w wieku reprodukcyjnym;
- niedobór witaminy D – suplementacja tej witaminy jest powszechna w Polsce, jednak aby była efektywna – co jest szczególnie istotne dla rozwoju płodu – musi być prowadzona pod kontrolą jej stężenia w surowicy. W przeciwnym wypadku możemy spotkać się z sytuacją, iż suplementacja powszechnie zalecanych dawek profilaktycznych (1000 – 2000 IU) nie wystarczy, i – pomimo zażywania leków – nadal występuje niedobór;
- podwyższone BMI;
- insulinooporność;
- ekspozycja na duże dawki jodu.
Jeśli u danej pacjentki występują ww czynniki, należy podjąć działania aby je wyeliminować i skorygować, częścią tego procesu powinny być ukierunkowane badania laboratoryjne.

Do czynników niemodyfikowalnych ryzyka dysfunkcji tarczycy należą:
- choroba autoimmunizacyjna – zwłaszcza, jeśli dotyczy tarczycy;
- zapalenia tarczycy;
- wole guzkowe;
- stan po leczeniu jodem radioaktywnym;
- stan po usunięciu tarczycy – tyreoidektomia.
Przeczytaj też:
- Tarczyca w ciąży – pod specjalnym nadzorem
- Niedobór hormonów tarczycy u kobiet w ciąży (Tarczyca w ciąży – pod specjalnym nadzorem cz.2)
Piśmiennictwo:
- Zhu i wsp.,Recent insights into the impact of immune dysfunction on reproduction in autoimmune thyroiditis,Clinical Immunology, Volume 224, 2021,108663, https://doi.org/10.1016/j.clim.2020.108663.
- Knøsgaard L, Andersen S, Hansen AB, Vestergaard P, Andersen SL. Thyroid function abnormalities and thyroid autoantibodies in Danish pregnant women. Clin Endocrinol (Oxf). 2020 Sep;93(3):329-338. doi: 10.1111/cen.14147. Epub 2020 Jan 22. PMID: 31876038.
- Bliddal S, Feldt-Rasmussen U, Rasmussen ÅK, Kolte AM, Hilsted LM, Christiansen OB, Nielsen CH, Nielsen HS. Thyroid Peroxidase Antibodies and Prospective Live Birth Rate: A Cohort Study of Women with Recurrent Pregnancy Loss. Thyroid. 2019 Oct;29(10):1465-1474. doi: 10.1089/thy.2019.0077. Epub 2019 Sep 24. PMID: 31407629.
- Karbownik-Lewinska M, Marcinkowska M, Stepniak J, Lewinski A. TSH ≥2.5 mIU/l is Associated with the Increased Oxidative Damage to Membrane Lipids in Women of Childbearing Age with Normal Thyroid Tests. Horm Metab Res. 2017 May;49(5):321-326. doi: 10.1055/s-0042-120712. Epub 2017 Apr 10. PMID: 28395379.