Poniższy artykuł jest zapisem webinaru (cz. I), który odbył się 23.05.2023 r.
Seronegatywne spondyloartropatie zapalne to grupa chorób, które charakteryzują się zapaleniem stawów. „Spondylo” oznacza, że może być zajęty kręgosłup, a „artropatie” to zaburzenia funkcjonowania stawów. Z kolei „seronegatywnymi” określa się je dlatego, że nie stwierdza się przy tej chorobie obecności czynnika reumatoidalnego w klasie IgM. W seronegatywnych spondyloartropatiach zapalnych najczęściej dochodzi do zajęcia różnych struktur kręgosłupa, takich jak krążki międzykręgowe, ale też stawów krzyżowo-biodrowych.
Seronegatywne spondyloartropatie zapalne są unikalną grupą chorób reumatologicznych, które charakteryzują się swoistymi dla siebie objawami i czynnikami etiologicznymi.
Do seronegatywnych spondyloartropatii zapalnych zaliczamy:
Choć każda z tych chorób ma swoje unikalne cechy, wszystkie mają wspólną element: zapalenie stawów.
Dlaczego należy leczyć seronegatywne spondyloartropatie zapalne?
Nieleczenie seronegatywnych spondyloartropatii zapalnych może prowadzić do poważnych konsekwencji, włączając niepełnosprawność, inwalidztwo, a nawet zgon. Powodem jest wysoki stan zapalny charakterystyczny dla tych chorób, który może doprowadzić do poważnych powikłań, w tym chorób układu sercowo-naczyniowego.
Przykładowo, nieleczone zesztywniające zapalenie stawów kręgosłupa (ZZSK) może prowadzić do przyspieszonej miażdżycy, zawału mięśnia sercowego i zgonu. Choć na pierwszy rzut oka główną przyczyną zgonu może wydawać się zawał mięśnia sercowego, rzeczywista przyczyna leży głębiej – prawdopodobnie zgon nie nastąpiłby, gdyby nie nieleczone ZZSK. Innymi słowy, ZZSK jest przyczyną, która inicjuje szereg zdarzeń prowadzących do przyspieszenia miażdżycy, udaru niedokrwiennego mózgu, zawału mięśnia sercowego i ostatecznie zgonu.
Postać osiowa i obwodowa seronegatywnych spondyloartropatii zapalnych
Spondyloartropatie zapalne o charakterze osiowym charakteryzują się przede wszystkim zajęciem kręgosłupa, z bólem i sztywnością pleców jako głównymi symptomami. Najczęstszym przykładem tej formy SpA jest spondylitis ankylopoetica (zespół Bechterewa), który może prowadzić do postępującego usztywnienia i zakrzywienia kręgosłupa.
Spondyloartropatie zapalne o charakterze obwodowym z kolei są związane z zajęciem stawów obwodowych, takich jak stawy kolanowe, biodrowe, łokciowe, czy skokowe. Objawami mogą być ból, obrzęk, a nawet utrata funkcji tych stawów. Przykładem choroby prezentującej głównie obwodowe zajęcie stawów jest psoriatyczne zapalenie stawów, które często występuje u osób z łuszczycą.
Choroby te różnią się od reumatoidalnego zapalenia stawów (RZS), które jest chorobą autoimmunologiczną charakteryzującą się symetrycznym zajęciem małych stawów, takich jak stawy śródręczno-paliczkowe czy śródstopno-paliczkowe. W przeciwieństwie do RZS SpA często prowadzą do asymetrycznego zajęcia stawów, zazwyczaj większych.
Ból charakterystyczny dla SpA
Ból w chorobach zapalnych stawów takich jak spondyloartropatie zapalne czy reumatoidalne zapalenie stawów (RZS) jest typowo zapalny. Charakteryzuje się on nasileniem rano (sztywność poranna) trwającym co najmniej godzinę, a często dłużej. To odróżnia go od chorób niezapalnych stawów, takich jak choroba zwyrodnieniowa stawów (OA), gdzie sztywność poranna zazwyczaj trwa mniej niż 30 minut.
W chorobach zapalnych, jak RZS czy spondyloartropatia, ból jest często zwiększany przez dotyk. Może to być oznaka obrzęku lub zapalenia w okolicach stawu. W przeciwieństwie do tego, w chorobach niezapalnych, takich jak OA, ból zazwyczaj nie jest nasilany przez dotyk, a pacjenci często opisują go jako ból „wewnątrz” stawu.
W przypadku pacjentów z spondyloartropatiami ból może występować w miejscach, gdzie ścięgna lub więzadła łączą się z kośćmi (entezy). Przykładem może być ból łokcia, który występuje, gdy pacjent opiera się na tym stawie. Ten rodzaj bólu, znanego jako entezopatia, jest charakterystyczny dla spondyloartropatii, takich jak spondylitis ankylopoetica.
Seronegatywne spondyloartropatie mogą mieć różnorodne objawy i dotyczyć różnych części ciała. Ich postać może być osiowa, obwodowa, czy mieszana, obejmująca zarówno kręgosłup, jak i stawy obwodowe. Przykładem objawów może być niesymetryczne zajęcie stawów krzyżowo-biodrowych i stawów obwodowych pod postacią daktylitis, czyli „kiełbaskowatych” palców.
Pacjenci mogą doświadczyć także objawów ogólnych, takich jak złe samopoczucie, przewlekłe zmęczenie, utrata masy ciała, gorączka, czy nocne poty. Seronegatywne spondyloartropatie mogą wystąpić w formie nieradiograficznej (zmiany niewidoczne na rentgenie, ale widoczne w rezonansie magnetycznym) lub radiograficznej.
W diagnostyce tych chorób używa się zarówno badań krwi, jak i badań obrazowych. Badania krwi obejmują oznaczanie czynnika reumatoidalnego (IgM RF) – jego brak jest typowy dla seronegatywnych spondyloartropatii – i antygenu HLA-B27, który jest obecny u większości pacjentów z tymi schorzeniami.
Warto jednak pamiętać, że nawet do 10% osób rasy kaukaskiej może mieć dodatni antygen HLA-B27 bez żadnej choroby. Dlatego, jeśli nie ma objawów sugerujących spondyloartropatię, nie jest zalecane samodzielne wykonanie tego badania. Istnieje także inny antygen –HLA-Cw6, który może być pomocny w diagnostyce łuszczycy i łuszczycowego zapalenia stawów, chociaż nie jest on wymieniany w kryteriach klasyfikacyjnych.
Miastenia (choroba Erba-Goldflama, ang. myasthenia gravis) to choroba rzadka, w całej Polsce z diagnozą mierzy się około 5 000 – 9 000 pacjentów, około 200 osób rocznie dowiaduje się o zachorowaniu. Z drugiej strony jest to poważny problem zdrowotny, z którego nie zdajemy sobie do końca sprawy. Pacjent – zanim otrzyma prawidłowe rozpoznanie – odwiedza wielu specjalistów (okulista, neurolog, logopeda, laryngolog, psychiatra), często pierwsza, błędna informacja brzmi, iż jest to zaburzenie psychiczne.
Czym jest miastenia?
Miastenia to choroba autoimmunologiczna, czyli schorzenie polegające na tym, iż organizm wytwarza przeciwciała przeciwko własnym strukturom. W miastenii są to przeciwciała przeciwko receptorom acetylocholiny, zlokalizowanym w zakończeniach nerwowych mięśni. W warunkach zdrowia acetylocholina wydzielana przez zakończenia nerwowe łączy się ze swoim receptorem w mięśniu, który dzięki temu dostaje impuls do skurczu. Jeśli są to mięśnie szkieletowe, człowiek może się poruszać, jeśli mięśnie oddechowe – oddychać, jeśli mięśnie żuchwy i gardła – można żuć pokarmy i je przełykać, jeśli mięśnie powiek – można otworzyć oczy. Natomiast jeśli receptor jest zablokowany, mięśnie nie mogą prawidłowo pracować, co pacjent odczuwa jako nadmierną męczliwość i osłabienie mięśni. W cięższych stadiach choroby osłabienie może prowadzić do pełnego bezwładu, dlatego kompletna nazwa choroby brzmi miastenia rzekomoporaźna (myasthenia gravis). Zwykle na początku wykonywania wysiłku mięśnie funkcjonują jeszcze prawidłowo, jednak bardzo szybko się męczą. W efekcie osoba chora zaczynając pewne czynności, nigdy nie wie, czy uda się je zakończyć. Pełną funkcjonalność pomagają przywrócić odpoczynek lub podanie odpowiednich leków.
Objawy miastenii
Jeśli zaburzenia funkcjonowania dotyczą mięśni szkieletowych, pacjent ma problemy z wykonywaniem codziennych czynności, takich jak zapinanie guzików, mycie zębów, czesanie się. Jego chód jest chwiejny, osoba chora zatacza się i wygląda jak po spożyciu alkoholu. Czasem pacjenci mówią, że czują się jak „szmaciane lalki”.
Nadmierna męczliwość mięśni twarzy skutkuje zaburzeniami mimiki, pacjent wygląda na osobę zmęczoną. Nawet jeśli się uśmiecha, jest to tzw. uśmiech Giocondy, czyli taki, przy którym nie podnoszą się kąciki ust (zmęczone mięśnie nie są w stanie unieść kącików do góry).
Zaburzenia funkcjonowania mięśni żwaczy to opadanie żuchwy, zaburzenia funkcjonowania mięśni odpowiedzialnych za proces żucia to problemy z gryzieniem, zaburzenia funkcjonowania mięśni gardła i krtani to problemy z połykaniem pokarmów oraz niewyraźna, nosowa i ściszająca się mowa.
Groźna postać choroby to zaburzenia funkcjonowania mięśni międzyżebrowych i przepony, co skutkuje trudnościami w oddychaniu, dusznością i w efekcie niewydolnością oddechową. Zdarza się wówczas, że chory musi być przyjęty do szpitala na oddział intensywnej terapii.
U ok. 70% pacjentów choroba zaczyna się jednak od zaburzeń funkcjonowania mięśni gałkoruchowych. Objawem ich dysfunkcji jest opadanie powieki, podwójne i nieostre widzenie.
Charakterystyczne dla miastenii jest nasilanie się objawów w ciągu dnia.
Kto najczęściej choruje na miastenię?
Miastenia może pojawić się w każdym wieku. Pierwszy szczyt zachorowań przypada na grupę osób w wieku 18-30 lat, 2-3 razy częściej dotyka kobiety. Następny szczyt pojawia się w grupie 45-60 lat, kobiety i mężczyźni chorują z taką samą częstotliwością. Jeśli natomiast choroba zaczyna się po 60 r.ż., statystycznie częściej będzie dotyczyła mężczyzn.
Niestety na miastenię chorują także dzieci. Pacjenci młodsi niż 16 lat, z dziecięcą postacią tej choroby, stanowią 10-16% wszystkich chorych.
Przyczyna choroby nie jest poznana, ale wiemy, że kluczowym czynnikiem w jej powstaniu jest grasica – gruczoł położony w klatce piersiowej, w którym powstają autoprzeciwciała. Grasica u osoby dorosłej jest w zaniku, ale 60% chorych na miastenię ma tzw. grasicę przetrwałą.
Miastenia nie jest uwarunkowana genetycznie, nie jest chorobą dziedziczną. Czasem pojawia się jednak rodzinnie – zwykle w tym samym pokoleniu, czyli wśród rodzeństwa lub kuzynostwa.
Diagnostyka miastenii
Miastenia może dotyczyć wielu obszarów ciała, dlatego czasem pacjenci muszą odwiedzić kilku specjalistów, aby poznać prawidłowe rozpoznanie.
W celu postawienia diagnozy lekarz zbiera wywiad i bada pacjenta. Elementem takiego badania jest ocena siły mięśniowej i męczliwości mięśni w czasie wykonywania pewnych czynności, np. wielokrotnego otwierania i zamykania oczu, czytania na głos lub liczenia.
Badania laboratoryjne w diagnostyce miastenii
przeciwciała przeciwko receptorowi acetylocholiny (anty-AChR, Ab-AChR) – obecność przeciwciał przeciwko AChR w stężeniu > 0,45 nmol/l potwierdza rozpoznanie choroby (miastenia seropozytywna). Niestety nie ma korelacji pomiędzy nasileniem objawów klinicznych a wysokością miana przeciwciał, badanie nie służy również do monitorowania leczenia. U ok. 15-20% chorych przeciwciała przeciwko receptorowi acetylocholiny w ogóle nie występują (miastenia seronegatywna).
Wykluczenie obecności obydwu typów przeciwciał nie wyklucza miastenii – istnieje niewielka grupa osób z miastenią „podwójnie seronegatywną” – bez anty-AChR i bez anty-MuSK.
Miastenia jest chorobąnieuleczalną, jednak stosowane dziś leczenie istotnie poprawia komfort życia chorego oraz skutkuje remisją. Lekami podstawowymi są leki antycholinoesterazowe, które łagodzą objawy i ułatwiają funkcjonowanie. Przyjmowanie leków trwa czasem kilka tygodniu lub nawet kilka miesięcy, zanim pacjent odczuje wyraźną poprawę. Jeśli stan chorego się nie poprawia, stosuje się leki immunosupresyjne lub glikokortykosteroidy. Wybór zastosowanych leków zależy od wielu czynników, m.in. odpowiedzi organizmu na leczenie, działań niepożądanych, chorób współistniejących, wieku, itp.
Jeśli miastenia spowodowana jest grasiczakiem, pacjent wymaga leczenia operacyjnego.
U osób z miastenią znacznie częściej występują zaburzenia depresyjne i lękowe. Obniżony komfort życia, upośledzenie funkcjonowania emocjonalnego oraz społecznego są wskazaniami do konsultacji ze specjalistami z zakresu psychologii lub psychiatrii i wdrożenia np. psychoterapii.
Piśmiennictwo:
MH Strugalska-Cynowska · 2008 — Miastenia i zespół miasteniczny Lamberta–Eatona (przydatność przeciwciał:przeciw AChR, titinie, MuSK i białkom kanału wapniowego
B. Emeryk-Szajewska Miastenia i zespół Lamberta-Eatona;immunopatologia, algorytmy diagnostyczne oraz skuteczność leczenia . Polski Przegląd Neurologiczny 2009; 5 (4):184–193
Termin „choroby tropikalne” nie został zdefiniowany przez Światową Organizację Zdrowia (World Health Organization, WHO), ale jest częścią słownictwa medycznego od XIX wieku.
Powstał w nieokreślonym czasie i stopniowo się konsolidował wraz z identyfikacją mikroorganizmów wywołujących te choroby i poznaniem mechanizmów ich przenoszenia.
W praktyce termin „choroby tropikalne” jest często rozumiany jako choroby zakaźne, które rozwijają się w gorących i wilgotnych warunkach. Do tych chorób zaliczamy, takie jednostki chorobowe jak na przykład: malaria, leiszmanioza, schistosomatoza, onchocerkoza (filarioza), filarioza limfatyczna, choroba Chagasa, trypanosomatoza afrykańska i denga.
Wiele organizmów, które stanowią podstawę chorób tropikalnych, to bakterie i wirusy z rozpoznaniem objawowym charakterystycznym dla tego typu zakażeń i tego typu organizmów. Mniej znane są te organizmy, które są bardziej złożone i określane jako pasożyty, malaria jest takim przykładem1.
Przyjmuje się, że ok. 15-20% turystów przebywających w strefie tropikalnej ma problemy zdrowotne podczas podróży, a 11% z nich cierpi na gorączkę o nieznanej przyczynie (FUO – Fever of Unknown Origin). 30-40% gorączek o nieznanym podłożu może mieć przyczynę zakaźną. Najczęstszymi przyczynami FUO związanymi z podróżami są: malaria, dur brzuszny, dury rzekome, żółta gorączka, wirusowe zapalenie wątroby (WZW), riketsjozy, ostra choroba retrowirusowa2.
Profilaktyka w podróży
Istotnym elementem dla osób podróżujących w obszary występowania „chorób tropikalnych” jest znajomość ich dróg przenoszenia w celu unikania ryzykownych zachowań, a w konsekwencji uniknięcia i zabezpieczenia człowieka przed narażeniem na zakażenie. Najistotniejsze drogi przenoszenia i czynniki zakaźne związane z możliwością ich zaistnienia to3:
Osoby udające się w podróż do regionów, w których epidemicznie czy też endemicznie występują „choroby tropikalne”, przed podróżą (najlepiej 2-3 miesiące) powinny skorzystać z poradnictwa ośrodków wyspecjalizowanych w tematyce medycyna podróży i choroby tropikalne w celu ustalenia przynajmniej kilku istotnych szczegółów poprawnego przygotowania się do tego przedsięwzięcia lub/i powinny skorzystać z ogólnie dostępnych poradników publikowanych na stronach instytucji do tego powołanych i akredytowanych.
Do czynności, które należy zaliczyć jako obowiązkowe przy planowaniu podróży, należą środki profilaktyczne dla podróżnych, takie jak3:
Konsultacja lekarska w celu odpowiedniego dobrania obowiązkowych i zalecanych szczepień ochronnych.
Ustalenie konieczności stosowania chemioprofilaktyki malarii w przypadku wyjazdu w rejony zagrożone tą chorobą.
Uzyskanie informacji dotyczących zasad nieswoistej profilaktyki chorób zakaźnych w tropiku oraz bezpiecznego zachowania w takiej podróży np. www.cdc.gov/travel lub www.iatatravelcentre.com.
Ustalenie wyposażenia apteczki podróżnej.
Uzyskanie informacji dotyczącej dostępności opieki zdrowotnej w rejonie docelowym.
Malaria jako przykład pasożytniczej choroby tropikalnej
Przyczyny malarii
Malaria (nazywana także zimnicą) to choroba pasożytnicza wywoływana przez zarodźce malarii. Wyróżnia się sześć gatunków zarodźców, które mogą zarażać człowieka i w rzeczywistości pięć pierwszych, tylko człowieka, i wywołać chorobę zwaną malarią (zimnicą). Należą do nich:
Plasmodium falciparum, zarodziec sierpowaty,
Plasmodium vivax, zarodziec ruchliwy,
Plasmodium ovale wallikieri, zarodziec owalny,
Plasmodium ovale curtisii, zarodziec owalny,
Plasmodium malariae, zarodziec pasmowaty,
Plasmodium knowlesi, zarodziec małpi.
Wektorem malarii jestkomar widliszek (Anopheles spp). Za większość przypadków ciężkiej malarii na świecie odpowiada zakażenie P. falciparum1. Wynika to z jego zdolności do zajmowania wszystkich stadiów erytrocytów (P. vivax i ovale atakuje wyłącznie retikulocyty i młode erytrocyty, a P. malariae starsze krwinki). Każdy z gatunków ma swój okres wylęgania: P. falciparum – 12 dni, P. vivax – 13 dni, P. ovale – 17 dni, P. malariae – 28 dni.
Przebieg malarii
Choroba ma przebieg związany z cyklem rozwojowym zarodźców i dzieli się na fazę przederytrocytalną i erytrocytalną. W fazie przederytrocytalnej po wniknięciu do organizmu sporozoity przemieszczają się do wątroby i atakują hepatocyty, w których przekształcają się w postać schizonta, dochodzi do ich namnożenia i uwolnienia następnej postaci, czyli merozoita, który ma zdolność wnikania do erytrocytów. W fazie erytrocytalnej dochodzi do namnażania trofozoitów i rozpadu erytrocytów, a także zainfekowania następnej partii krwinek czerwonych. Namnażanie i rozpad erytrocytów przyjmują postać synchronizowaną czasowo z objawami cyklicznej gorączki. Cykle są różne dla różnych gatunków Plasmodium spp i wynoszą dla P. vivax i P. ovale 48 h [trzeciaczka], dla P. malariae i P. falciparum 72 h [czwartaczka] i dla P. knowlesi 24h. Po kilku lub kilkunastu cyklach powstają gametocyty, które są inwazyjne dla komarów. Podczas żerowania komary zarażają się, a w ich organizmach zachodzi faza płciowa cyklu rozwojowego.11,13
Najbardziej zjadliwym pasożytem jest P. falciparum, odpowiada on za prawie 90% zgonów, z kolei P. vivax odpowiada za prawie 80% zachorowań.
Malaria – statystyki
Według raportu WHO liczba zgonów z powodu malarii (zimnicy) z roku na rok jest coraz mniejsza. W 2000 roku na świecie w wyniku malarii zmarło około miliona osób, natomiast w 2017 roku liczba ta zmniejszyła się do wartości poniżej 450 000, czyli o więcej niż 50%.4,6
W podanej powyżej liczbie zgonów około 2/3 (290 000) dotyczyło dzieci poniżej 5. roku życia, co daje średnio 800 zgonów najmłodszych dzieci dziennie.5
Natomiast w ostatnim raporcie Europejskiego Centrum ds. Zapobiegania i Kontroli Chorób (ECDC) z 2023 roku odnoszącego się do stanu epidemiologii malarii w 2020 roku na świecie wskazano, że oszacowano około 241 milionów przypadków infekcji w 85 endemicznych krajach z około 627 tysiącami zgonów. Do transmisji malarii dochodziło głównie na dużej części obszarów centralnej i południowej Ameryki, Afryki, Azji i Oceanii. Można z tych dany wywnioskować, że tendencja spadkowa została zahamowana, a nawet doszło do jej odwrócenia i ponownego wzrostu liczby zgonów.7,8,9
Ten sam raport podaje sytuację epidemiologiczną w krajach Unii Europejskiej w latach 2016 -2020. W tym okresie w 2016 roku stwierdzono 8226 przypadków malarii, a w 2020 roku tylko 2369, w Polsce zaraportowano 38 przypadków w 2016 roku i tylko 7 przypadków w 2020 roku. Taka sytuacja była prawdopodobnie spowodowana dwoma głównymi czynnikami, pierwszym związanym z profilaktyką, prewencją i wzrastającą świadomością podróżujących Europejczyków, drugi z sytuacją pandemiczną związaną z wirusem SARS-CoV-2, która radykalnie ograniczyła turystykę do strefy tropikalnej i subtropikalnej.7,8,9
Profilaktyka malarii
Profilaktyka malarii (zimnicy) została zdefiniowana przez WHO jako strategia ABCDE, gdzie skrót nazwy pochodzi od pierwszych liter angielskich nazw działań składających się na prawidłowe działania profilaktyczne.6,10 Działania te to:
A. Świadomość ryzyka (edukacja) – Awareness
B. Ochrona przed ukłuciami komarów – Bites of mosquitoes
C. Chemioprofilaktyka – Chemoprophylaxis
D. Rozpoznanie (okres od 7 dni pobytu do 3 miesięcy po powrocie) – Diagnosis
E. Unikanie aktywności w środowisku sprzyjającym ekspozycji (np. tereny podmokłe) – Environments
Z kolei zwalczanie malarii (zimnicy) można określić jako proces wielopoziomowy i wielokierunkowy. Na który składają się takie działania jak:3,4,10
profilaktyka nieswoista (stosowanie repelentów i moskitier nasączonych środkami owadobójczymi),
prace nad modyfikacją genetyczną gatunków komarów przenoszących zimnicę,
stosowanie szczepień, szczególnie dzieci w obszarach endemicznych,
profilaktyka swoista (np. chemioprofilaktyka, czyli stosowanie leków przeciwmalarycznych przed podróżą do krajów endemicznych i w jej trakcie).
WHO nie zaleca podróży na tereny endemiczne z niemowlętami i małymi dziećmi ze względu na ryzyko ciężkiego przebiegu i powikłań malarii wywołanej przez P. falciparum, a jeżeli jest to konieczne, bezwzględnie należy przestrzegać zasad profilaktyki.10
WHO podaje, jakie działania (zalecenia) profilaktyczne należy zastosować w zależności od ryzyka, jakie niesie za sobą podróż w określone regiony endemiczne występowania malarii.6,10
Zapobieganie malarii w podróży w zależności od ryzyka zakażenia i lekooporności zarodźców malarii:
Samokontrola po podróży w tropiki
Dla malarii nie ma typowych dolegliwości chorobowych, szczególnie w początkowej fazie choroby, dlatego początkowe objawy u zarażonego turysty są zwykle niecharakterystyczne i można je pomylić z grypą lub innymi infekcjami wirusowymi. Jeżeli po powrocie z podróży z obszaru gdzie endemicznie występuje malaria, wystąpią objawy grypopodobne lub wystąpią takie objawy jak gorączka z dreszczami oraz potami, ból głowy i mięśni, osłabienie i złe samopoczucie i dodatkowo można te objawy skojarzyć z ukąszeniem przez komara, to powinno się skorzystać z konsultacji lekarskiej w placówce zajmującej się diagnozowaniem chorób tropikalnych. Konieczność skorzystania z konsultacji znacznie wzrasta, gdy dodatkowo wystąpią nudności, wymioty, biegunki i żółtaczka. Należy pamiętać, że każdy dzień zwłoki niesie za sobą ryzyko ciężkiego przebiegu choroby.14
Jeżeli zamiast do specjalistycznej jednostki pacjent z w/w objawami trafi do lekarza POZ, to powinien pamiętać, by koniecznieprzekazać informację o swoim pobycie w krajach strefy tropikalnej, nawet jeśli od pobytu upłynęło kilka miesięcy.
Podsumowanie
Ekspozycja na zarodźce malarii nie zapewnia ochrony przed ponownym zachorowaniem, a nabyta w ten sposób odporność jest tylko częściowa.
Od dawna podejmowane są próby opracowania skutecznej szczepionki przeciwko zarodźcom wywołującym malarię. Ze względu na to, że Plasmodium spp. to organizm eukariotyczny o bardzo złożonym cyklu życiowym wywołującym zróżnicowaną odpowiedź immunologiczną w organizmie człowieka, prace nad większością szczepionek kończyły się niepowodzeniem.
Ostatnio ukończono jednak prace nad nową szczepionką przeciwko malarii (zimnicy), która ma szansę wpłynąć – przynajmniej częściowo – na statystykę zachorowań na malarię wywoływaną przez Plasmodium falciparum.11,12
Szczepionka ta to RTS,S/AS01 – jest ona jedyną szczepionką przeciwko malarii (zimnicy), która ukończyła III fazę badań klinicznych. Jest to szczepionka podjednostkowa, zawierająca białko PfCSP (Plasmodium falciparum circumsporozoite protein) obecne na powierzchni sporozoitów. Nie zakłada się, że stosowanie tej szczepionki przyczyni się do całkowitej eradykacji zachorowań wywołanych przez Plasmodium falciparum. Prawdopodobnie nie będzie ona również odgrywać istotnej roli w medycynie podróży – przemawia za tym jej ograniczona skuteczność oraz konieczność stosowania wielu dawek w stosunkowo długim przedziale czasu. Na razie ta szczepionka jest przeznaczona wyłącznie do stosowania u osób zamieszkujących Afrykę.
Informacje dodatkowe:
Laboratorium Analiz Lekarskich ALAB Gdynia Powstania Styczniowego 9b, świadczące usługi medycyny laboratoryjnej dla Uniwersyteckiego Centrum Medycyny Morskiej i Tropikalnej wykonuje jako jedyne medyczne laboratorium diagnostyczne pełen pakiet badań z zakresu diagnostyki malarii.
Piśmiennictwo:
Pujara P et al, An introduction to Tropical Disease: A review article. International Journal of Medical Microbiology and and Tropical Diseases 2016; 2(3): 81-83
European Centre for Disease Prevention and Control (ECDC). Introduction to the Annual Epidemiological Report [internet]. Stockholm: ECDC; 2017 [cited 14 October 2022]. Available from: http://ecdc.europa.eu/annual-epidemiological-reports/methods
Poniższy artykuł jest zapisem webinaru, który odbył się 26.04.2023 r.
Choroby układowe tkanki łącznej to zróżnicowana grupa chorób, charakteryzujących się wysokim procesem zapalnym i mających podłoże immunologiczne. Mówimy tu o stanach, w których układ immunologiczny ma kluczowe znaczenie w powstawaniu tych chorób.
Dlaczego choroby tkanki łącznej nazywa się układowymi?
Termin „układowe” w nazwie tych chorób wskazuje, że choroby te mogą dotyczyć wielu różnych układów naszego ciała. Mogą one dotyczyć skóry, układu krwionośnego, krwiotwórczego, sercowo-naczyniowego, oddechowego, pokarmowego, narządu wzroku czy układu nerwowego. Każdy układ w naszym organizmie może być „zajęty” przez te choroby.
Układowe choroby tkanki łącznej są bardzo poważne i przewlekłe. Chociaż są nieuleczalne, dąży się do obniżenia aktywności choroby, czyli do jej remisji. Podział tych chorób jest obszerny, ale w tym artykule skupimy się na tych najpowszechniejszych.
Podstawowe choroby tkanki łącznej
Do podstawowych chorób tkanki łącznej zaliczamy:
toczeń rumieniowaty układowy,
twardzinę układową,
zespół Sjögrena,
idiopatyczne zapalenie mięśni,
mieszaną chorobę tkanki łącznej,
polimialgię reumatyczną.
Wszystkie te choroby, choć występują rzadko, są bardzo poważne i wymagają szybkiego diagnozowania i włączenia skutecznego leczenia na wczesnym etapie, aby osiągnąć remisję i uniknąć powikłań.
Struktura tkanki łącznej – macierz zewnątrzkomórkowa. Tkanka łączna podtrzymuje i chroni inne tkanki i narządy organizmu człowieka.
Toczeń rumieniowaty układowy
Szczegółowe informacje na temat tocznia rumieniowatego – charakterystyki, objawów i diagnostyki znajdziesz w artykule poświęconym tej chorobie.
Twardzina układowa – co to jest?
Twardzina układowa to ciężka przewlekła choroba autoimmunologiczna, która charakteryzuje się twardnieniem i zwłóknieniem skóry oraz narządów wewnętrznych, takich jak płuca, serce, nerki i przewód pokarmowy. Choroba ta jest bardzo poważna i może skracać życie. Występuje ona trzy do cztery razy częściej u kobiet, a najczęściej diagnozowana jest między 30. a 50. rokiem życia.
Chociaż istnieją metody leczenia, to koncentrują się one na leczeniu objawów specyficznych dla danego narządu, który jest aktualnie zajęty przez chorobę. Takie leczenie określa się jako leczenie „narządowo-swoiste”.
Twardzina układowa ma różne postaci. Występuje postać ograniczona, która zazwyczaj początkowo dotyka tylko skórę, ale po kilku latach może rozszerzyć się na inne narządy, takie jak płuca, przewód pokarmowy, nerki i serce. Proces ten jest jednak powolny i rozłożony w czasie.
Drugą postacią jest twardzina układowa uogólniona, która jest bardziej agresywna. W tej formie choroby, zajęcie skóry i narządów wewnętrznych, takich jak nerki, układ oddechowy, płuca, serce i przewód pokarmowy, następuje szybko. Ta postać choroby może znacząco wpływać na funkcjonowanie pacjenta.
Twardzina układowa – przyczyny i objawy
Twardzina układowa charakteryzuje się postępującym włóknieniem skóry i narządów wewnętrznych. Do procesu włóknienia prowadzą zaburzenia w funkcjonowaniu komórek zwanych fibroblastami. Fibroblasty są komórkami odpowiedzialnymi za produkcję kolagenu i innych białek strukturalnych, które tworzą szkielet naszej tkanki łącznej. Kiedy te komórki nie funkcjonują prawidłowo, może dojść do nadmiernego tworzenia tkanki łącznej, prowadząc do zgrubienia i stwardnienia skóry oraz narządów wewnętrznych.
Jednym z najbardziej charakterystycznych objawów twardziny układowej jest objaw Raynauda. Jest to stan, w którym niewielkie naczynia krwionośne w skórze skurczają się jako reakcja na zimno lub stres, powodując bladość, sinienie i czerwoność palców rąk lub stóp. Ten objaw jest bardzo częsty w twardzinie układowej, pojawiając się prawie u wszystkich pacjentów z uogólnioną formą choroby i u większości pacjentów z ograniczoną formą choroby.
Twardzina układowa może prowadzić do zajęcia różnych narządów, ale najpoważniejsze jest zajęcie płuc. Choroba śródmiąższowa płuc, która jest częstą komplikacją twardziny, może prowadzić do pogorszenia tolerancji na wysiłek, suchego kaszlu, problemów ze snem i duszności. Ta choroba płuc jest rozpoznawana poprzez badania obrazowe, takie jak tomografia komputerowa wysokiej rozdzielczości, która może wykazać charakterystyczne zmiany w płucach związane z włóknieniem śródmiąższowym.
Rozwój twardziny układowej
Rozwój twardziny układowej może obejmować stopniowe pogorszenie funkcji wielu różnych narządów. W szczególności choroba śródmiąższowa płuc może prowadzić do włóknienia płuc. Włóknienie to proces, w którym zdrowa tkanka płuc zostaje zastąpiona przez bliznowate tkanki, które nie są w stanie efektywnie wymieniać tlenu. W rezultacie, pacjenci z włóknieniem płuc mogą doświadczać duszności i zmniejszonej tolerancji na wysiłek.
Twardzina układowa może również wpływać na stawy, prowadząc do bólu i ograniczenia ruchomości. Zajęcie przewodu pokarmowego jest również częstym objawem twardziny układowej, z najczęstszymi objawami obejmującymi trudności w przełykaniu i problemy z przewodem pokarmowym.
Twardzina układowa może również prowadzić do poważnych problemów z nerkami, znanych jako twardzinowy przełom nerkowy. W takim przypadku nerki przestają prawidłowo funkcjonować, co prowadzi do gwałtownego wzrostu ciśnienia krwi. Twardzinowy przełom nerkowy jest stanem zagrożenia życia i wymaga natychmiastowej interwencji medycznej.
Leczenie twardziny układowej
Leczenie twardziny układowej wymaga dostosowania do indywidualnego przebiegu choroby i objawów u pacjenta. Jako że twardzina układowa jest chorobą autoimmunologiczną, cel leczenia polega na kontrolowaniu nadaktywnego systemu immunologicznego i łagodzeniu objawów.
Podstawowe leki stosowane w leczeniu twardziny układowej to leki przeciwzapalne i immunosupresyjne. Wybór leku zależy od objawów i zaangażowania poszczególnych narządów. W leczeniu twardziny układowej często stosuje się leki takie jak metotreksat, cyklofosfamid i mykofenolan mofetylu. Leki te pomagają zmniejszyć objawy zapalenia i spowolnić postęp choroby.
Istnieje również nowy, obiecujący lek przeciw włóknieniu – Nintedanib. Może on być pomocny w leczeniu twardziny układowej z zajęciem płuc. Leczenie nerek może obejmować inhibitory konwertazy angiotensyny, które są stosowane do leczenia wysokiego ciśnienia krwi. W przypadku objawów skórnych skuteczne mogą być leki takie jak metotreksat, cyklosporyna, a także leki przeciwmalaryczne.
Zapalenia skórno-mięśniowe i wielomięśniowe – objawy
Zapalenie skórno-mięśniowe i wielomięśniowe to rodzaje miopatii zapalnych, czyli chorób, które atakują mięśnie.
Zapalenie wielomięśniowe (polymyositis) charakteryzuje się postępującym osłabieniem mięśni, zwłaszcza mięśni proksymalnych, czyli tych, które są bliższe środkowi ciała. Zwykle obejmuje to mięśnie karku, ramion, ud oraz obręczy miednicznej.
Zapalenie skórno-mięśniowe (dermatomyositis), z drugiej strony, to stan, który dołącza do objawów zapalenia wielomięśniowego różne objawy skórne. Objawy te mogą obejmować:
Rumień heliotropowy: to fioletowe przebarwienie wokół oczu, które może być bardzo charakterystyczne dla zapalenia skórno-mięśniowego, zwłaszcza jeśli towarzyszy mu obrzęk.
Objaw Szala: zaczerwienienie skóry w kształcie litery V.
Objaw Kabury: zaczerwienienie skóry wokół obręczy miednicznej.
Objaw Gottrona: zasinienie na stawach, szczególnie na stawach rąk, między paliczkami i śródręczem.
Grudki Gottrona: grudki, które mogą występować w tych samych miejscach, gdzie występuje objaw Gottrona.
Podstawowym objawem obu tych stanów jest postępujące osłabienie mięśni. Chorzy mogą mieć problemy z poruszaniem się, na przykład ze wstaniem z krzesła czy podnoszeniem przedmiotów. Ważne jest, że to osłabienie nie jest spowodowane bólem – to jest znacząca różnica między tymi stanami a innymi chorobami mięśni, takimi jak polimialgia reumatyczna, która powoduje silny ból.
Oba te stany są rzadkie i najczęściej występują w dwóch grupach wiekowych: między 15. a 35. rokiem życia oraz między 60. a 65. rokiem życia. W zapaleniu skórno-mięśniowym, obecność zmian skórnych jest dodatkowym objawem dołączonym do osłabienia mięśni obserwowanego w zapaleniu wielomięśniowym.
Diagnostyka i leczenie zapalenia skórno-mięśniowego i zapalenia wielomięśniowego
Diagnostyka zapalenia skórno-mięśniowego i zapalenia wielomięśniowego obejmuje szereg badań.
Podstawowe badania krwi na obecność markerów uszkodzenia mięśni, takich jak ASPAT, ALAT, LDH, CPK (kinaza fosfokreatynowa) i aldolaza, mogą sugerować uszkodzenie mięśni i podniesienie tych parametrów może sugerować obecność choroby. Należy jednak pamiętać, że nie wszystkie osoby z tymi chorobami będą miały podwyższone te parametry.
Badane mogą też być przeciwciała przeciwjądrowe (ANA). Specyficzne dla tych chorób są przeciwciała anty-Jo-1 (charakterystyczne dla zespołu antysyntetazowego i zapalenia wielomięśniowego) oraz anty-Mi-2 (charakterystyczne dla zapalenia skórno-mięśniowego). Obecność tych przeciwciał w surowicy pacjenta może sugerować obecność tych chorób, ale nie jest to wystarczające do postawienia diagnozy.
Najdokładniejszym sposobem rozpoznania tych chorób jest jednak biopsja skórno-mięśniowa, gdzie fragment mięśnia i skóry jest pobrany i badany pod mikroskopem. Jednym z kryteriów rozpoznania jest obecność nacieku limfocytarnego w mięśniach.
Innym badaniem diagnostycznym jest badanie elektromiograficzne (EMG). Może ono wykazać uszkodzenie mięśni. Z kolei rezonans magnetyczny z kontrastem może pokazać, które mięśnie są najbardziej zapalone. Te dwa badania mogą pomóc lekarzowi zdecydować, skąd pobrać wycinek do biopsji skórno-mięśniowej.
Leczenie tych chorób zazwyczaj obejmuje glikokortykosteroidy oraz leki immunosupresyjne, które modyfikują przebieg choroby.
Zapalenie skórno-mięśniowe i wielomięśniowe a nowotwory
Podczas monitorowania leczenia zapalenia skórno-mięśniowego i zapalenia wielomięśniowego, konieczne jest zwrócenie szczególnej uwagi na potencjalne powiązane ryzyko nowotworów. Osoby z tą chorobą mają zwiększone ryzyko wystąpienia różnych typów nowotworów. W przypadku zapalenia wielomięśniowego (polimiozytis), ryzyko nowotworu wzrasta dwukrotnie, podczas gdy zapalenie skórno-mięśniowe (dermatomiozytis) zwiększa to ryzyko sześciokrotnie w porównaniu do populacji ogólnej.
Wśród nowotworów, na które należy zwrócić szczególną uwagę, sąrak jajnika, rak pęcherza moczowego, rak piersi, rak płuc oraz rak żołądka. W związku z tym, osoby z tą chorobą powinny poddawać się regularnym badaniom, które mogą pomóc we wczesnym wykryciu tych nowotworów.
Mieszana choroba tkanki łącznej – charakterystyka
Mieszana choroba tkanki łącznej (ang. Mixed Connective Tissue Disease, MCTD) to rzadka choroba autoimmunologiczna, która charakteryzuje się objawami i cechami kilku innych chorób autoimmunologicznych, w tym reumatoidalnego zapalenia stawów (RZS), tocznia rumieniowatego układowego (SLE), twardziny układowej (SSc) i idiopatycznych miopatii zapalnych.
Pacjenci z MCTD mogą doświadczać wielu objawów związanych z tymi chorobami, takich jak:
Symetryczne zajęcie stawów, szczególnie małych stawów stóp i rąk. Ból, obrzęk i sztywność poranna trwająca często ponad godzinę są często obserwowane.
Osłabienie mięśni i zmiany skórne związane z idiopatycznymi miopatiami zapalnymi.
Charakterystyczne dla MCTD jest występowanie przeciwciała U1 RNP w bardzo wysokim mianie. Przeciwciało to jest częścią tzw. przeciwciał przeciwjądrowych i jest specyficzne dla MCTD. Wykrywa się go za pomocą badania ANA (przeciwciała przeciwjądrowe) z użyciem wysokich rozcieńczeń, często zaczynając od 1 do 1280, a czasem dochodząc nawet do 1 do 5120.
Leczenie mieszanej choroby tkanki łącznej
Leczenie MCTD zależy od dominującej choroby, czyli od tego, które objawy są najbardziej wyraźne i sprawiają najwięcej problemów pacjentowi. Leczenie może obejmować:
metotreksat, jeśli dominuje RZS,
leki przeciwmalaryczne, takie jak Chlorochina lub Hydroksychlorochina, jeśli dominuje SLE,
wysokie dawki glikokortykosteroidów, jeśli dominuje idiopatyczna miopatia zapalna,
cyklofosfamid, mykofenolan mofetylu lub nintedanib, jeśli dominuje twardzina układowa, zwłaszcza z zajęciem płuc.
Ważne jest indywidualne dopasowanie leczenia do dominujących objawów i zespołów chorobowych. Monitorowanie choroby i dostosowanie terapii powinno być przeprowadzane regularnie w celu optymalizacji leczenia i kontroli objawów.
Zespół Sjögrena
Szczegółowe informacje na temat zespołu Sjögrena – charakterystyki, objawów i diagnostyki znajdziesz w artykule poświęconym tej chorobie.
Polimialgia reumatyczna – charakterystyka
Polimialgia reumatyczna jest chorobą reumatyczną, która najczęściej dotyka osoby starsze, zwłaszcza te po 50. lub 60. roku życia. To jest choroba, która jest związana z bólem i sztywnością w dużych mięśniach ciała, zwłaszcza tych wokół ramion, szyi i bioder. W przeciwieństwie do niektórych innych chorób mięśniowych ból i sztywność nie są spowodowane osłabieniem mięśni, ale raczej bólem mięśni.
Typowe objawy to ból i sztywność ramion, szyi i bioder, które mogą być na tyle silne, że utrudniają ruchy takie jak podnoszenie ramion lub wstawanie. Często pacjenci doświadczają ogólnego obrzęku, w tym „puffy fingers” – obrzęk całej ręki, a nie tylko stawów, co jest typowe dla innej choroby zwaną EORA (elderly onset rheumatoid arthritis), czyli reumatoidalne zapalenie stawów występujące u osób starszych. Ta choroba musi być różnicowana z polimialgią reumatyczną.
Inne objawy, które mogą wystąpić w polimialgii reumatycznej, to bardzo duże zmęczenie, utrata masy ciała, wysoka gorączka bez wyraźnej przyczyny. Wielu pacjentów doświadcza także bardzo wysokich wskaźników stanu zapalnego, takich jak OB i CRP.
Polimialgia reumatyczna jest również związana z wyższym ryzykiem występowania nowotworów, dlatego pacjenci z tą diagnozą powinni przejść badania przesiewowe w celu wykrycia ewentualnego nowotworu.
Co więcej, polimialgia reumatyczna może współwystępować z chorobą Hortona, która jest olbrzymiokomórkowym zapaleniem tętnic, w tym tętnicy skroniowej. To stanowi jedną z nagłych sytuacji w reumatologii, ponieważ może prowadzić do utraty wzroku. Aby zapobiec temu powikłaniu, pacjenci muszą otrzymywać bardzo wysokie dawki glikokortykosteroidów.
Podsumowanie – pierwsze objawy układowych chorób łącznych
Układowe choroby łącznej mogą dawać bardzo charakterystyczne objawy. Zanim jednak się rozwiną, widoczne mogą być sygnały, które mogą sugerować rozwijającą się chorobę. Do konsultacji z lekarzem powinny skłonić:
przewlekłe zmęczenie,
epizody gorączki lub stanu podgorączkowego, które nie mogą być wyjaśnione infekcją, czy inną chorobą,
niezamierzona utrata masy ciała, bez zmiany stylu życia czy diety,
nocne poty, niezwiązane z warunkami zewnętrznymi (takimi jak wysoka temperatura otoczenia).
Niegdyś układowe choroby tkanki łącznej nazywane były „kolagenozami”. Było to mylące, ponieważ sugerowało, że te choroby są związane tylko z kolagenem. Obecnie wiadomo, że te choroby mają znacznie szersze podłoże.
Gdy pada hasło „alergia”, często myślimy o rodzaju reakcji, który znamy doskonale z filmów czy seriali. Scena zazwyczaj przebiega następująco: w posiłku spożywanym przez bohatera przypadkowo (lub celowo) znajduje się produkt, na który bohater ten ma silną alergię. Niemalże natychmiast po kontakcie z alergenem zaczynają pojawiać się niepokojące objawy: obrzęk, zaczerwienienie, wysypka, trudności w oddychaniu. Tak przedstawiane reakcje na pokarmy upodobali sobie szczególnie twórcy seriali komediowych, takich jak Przyjaciele (alergia na kiwi), Jak Poznałem Waszą Matkę (alergia na orzechy ziemne i skorupiaki), Współczesna Rodzina (alergia na soję). Reakcja przebiegająca w ten sposób to właśnie alergia IgE-zależna. Przedstawienie jej na filmowym ekranie jako coś zabawnego to zabieg dosyć niefortunny, ponieważ jest to najniebezpieczniejsza forma alergii, mogąca stanowić zagrożenie życia.
Czym jest alergia IgE-zależna?
Alergia IgE-zależna to forma immunologicznej nadwrażliwości na określone alergeny pokarmowe i/lub wziewne, związana z nadmierną produkcją przeciwciał IgE (są to tzw. swoiste przeciwciała klasy IgE). Cechą charakterystyczną tej formy alergii jest szybkość reakcji: objawy często występują natychmiast po kontakcie z alergenem, choć mogą być także opóźnione do 4 godzin. Reakcja może przybrać różne formy: od wysypki, obrzęku, kaszlu, kichania, biegunki aż do reakcji ogólnoustrojowej, prowadzącej do znacznego obniżenia ciśnienia tętniczego krwi – czyli wstrząsu anafilaktycznego. To właśnie ryzyko anafilaksji sprawia, że alergie IgE-zależna są tak niebezpieczne.
Jak działa alergia IgE-zależna?
U osoby predysponowanej do rozwoju alergii pierwszy kontakt z alergenem powoduje wytworzenie przeciwciał IgE, bez wystąpienia żadnych objawów. Wytworzone przeciwciała IgE otaczają komórkę tuczną (jedna z komórek układu odpornościowego) i czekają w gotowości na kolejny kontakt z alergenem. Gdy to nastąpi, przeciwciała łączą się z alergenem i dochodzi do degranulacji („pęknięcia”) komórki tucznej. W efekcie zostają uwolnione substancje, odpowiadające za wystąpienie reakcji alergicznej, zwłaszcza histaminy. Dlatego też skuteczne łagodzenie objawów alergii zazwyczaj opiera się na stosowaniu leków przeciwhistaminowych.
Które alergeny najczęściej powodują alergie IgE-zależne?
Najczęstsze alergeny prowadzące do wystąpienia alergii IgE-zależnych znajdziemy w produktach mlecznych, jajach, orzeszkach ziemnych, orzechach, rybach, skorupiakach, soi, pszenicy czy sezamie. Mechanizm IgE-zależny odpowiada także za większość reakcji na pyłki drzew i traw oraz towarzyszących im alergii krzyżowych (zespół pyłkowo-pokarmowy).
Na szczególną uwagę zasługują alergeny orzechów arachidowych (Ara h 1, Ara h 2, Ara h 3, Ara h 6, Ara h 9), mleka (kazeina), jajka (owomukoid) i pszenicy (Tri a 19), ponieważ to właśnie one często odpowiadają za wystąpienie ciężkich reakcji alergicznych, w tym wstrząsu anafilaktycznego. Warto zaznaczyć, że alergia IgE-zależna może powstać na dowolne białko pokarmowe, choć wcześniej wymienione alergeny powodują ten rodzaj nadwrażliwości najczęściej (należą do wielkiej 9 alergenów pokarmowych zaproponowanych przez FDA – Food and Drug Administration).
Diagnostyka alergii IgE-zależnych
Pierwszym krokiem przed rozpoczęciem diagnostyki jest przeprowadzenie dokładnego wywiadu z pacjentem w celu wstępnej oceny, z jakim rodzajem i formą reakcji mamy do czynienia. Przy podejrzeniu występowania alergii IgE-zależnej najczęściej początkiem diagnostyki są testy skórne i/lub testy alergologiczne z krwi.
Testy skórne uogólniając, polegają na nakłuciu skóry i wprowadzeniu alergenu, a następnie obserwowaniu reakcji skórnej, takiej jak zaczerwienienie, świąd lub obrzęk; wszystko pod okiem lekarza, alergologa. Testy alergiczne z krwi polegają na badaniu poziomu swoistych przeciwciał IgE skierowanych przeciwko określonym alergenom. W ramach testów z krwi zazwyczaj przesiewowo wykonuje się badanie panelowe, w kierunku alergii wziewnych i/ lub pokarmowych z oznaczeniem swoistych przeciwciał IgE. Możemy wykonać również oznaczanie całkowitego stężenia IgE we krwi, które w alergiach często jest podwyższone. Jednakże wzrost całkowitego IgE we krwi może również występować w innych stanach, takich jak choroby pasożytnicze, zakażenia bakteryjne i wirusowe, sarkoidoza, aspergiloza płucna, łuszczyca, stany niedoboru odporności i inne.
Doskonalszą metodą diagnostyczną w przypadku alergii IgE-zależnych jest diagnostyka molekularna. Metoda ta pozwala nam nie tylko ocenić, które czynniki powodują alergie (np. mleko), ale także, które dokładnie białka w nich zawarte są źródłem reakcji (np. kazeina).
W przypadku alergii pokarmowych wiedza te jest potrzebna, aby określić, czy alergen może być spożywany po obróbce termicznej, czy też musi być wyeliminowany w każdej postaci. Przykładowo, przy alergii na kazeinę produkty mleczne muszą być wyeliminowane w każdej postaci, natomiast przy alergii na białka serwatki produkty po odpowiedniej obróbce termicznej często nie powodują objawów.
Co więcej, diagnostyka molekularna pozwala nam ustalić, jak duże jest ryzyko wystąpienia ciężkiej reakcji na alergen – w tym wstrząsu anafilaktycznego. Przykładowo, alergia na białko orzecha ziemnego Ara h 8 niezwykle rzadko powoduje ciężkie reakcje alergiczne. Co więcej, białko to traci właściwości alergizujące po obróbce termicznej. Tymczasem alergia na białka zapasowe orzechów ziemnych (Ara h 1, Ara h 2, Ara h 3 i Ara h 6) wiąże się z wysokim ryzykiem anafilaksji. Co gorsza, obróbka termiczna nie ma na nie wpływu. Niezwykle ciekawym przypadkiem jest także alergia na białko Ara h 9, która może wiązać się ze wstrząsem anafilaktycznym, ale zazwyczaj w towarzystwie dodatkowego czynnika wyzwalającego, jakim może być intensywny wysiłek fizyczny, silny stres, infekcja czy stosowanie używek.
Z kolei w przypadku alergii na pyłki diagnostyka molekularna jest niezwykle pomocna w rozpoznaniu alergii krzyżowych z pokarmami, które pozostają nieuchwytne w klasycznych badaniach swoistych przeciwciał IgE, czy w kwalifikacji do swoistej immunoterapii.
Jeśli to nie alergia IgE-zależna… to jaka?
Musimy pamiętać, że poza alergiami IgE-zależnymi występują także alergie niezależne od IgE.
Zgodnie z ustaleniami Coombsa i Gella z 1963 roku w zależności od rodzaju komórek biorących udział w reakcji alergicznej rozróżnia się cztery główne typy nadwrażliwości:
I typ nadwrażliwości – reakcja natychmiastowa zależna od przeciwciał IgE
W ten typ reakcji zaangażowane są limfocyty B, komórki tuczne (mastocyty) i bazofile. Ten rodzaj alergii jest najbardziej powszechny i dotyczy ok. 35% społeczeństwa.
II typ nadwrażliwości – reakcja cytotoksyczna, zależna od przeciwciał IgM i IgG
W ten typ nadwrażliwości zaangażowane są komórki NK (natural killer) i makrofagi. Przeciwciała błędnie wiążą się z komórkami organizmu i powodują ich niszczenie, w wyniku aktywacji innych mechanizmów układu odpornościowego takich jak np. układ dopełniacza czy komórek NK. Przykładem choroby z udziałem tego mechanizmu jest pokrzywka przewlekła.
Typ III – reakcja kompleksów immunologicznych
W ten typ reakcji zaangażowane są neutrofile i płytki krwi. Przeciwciała IgG przyłączają się do antygenów, tworząc tzw. kompleksy immunologiczne. Kompleksy te odkładają się w tkankach i aktywują białka tzw. układu dopełniacza. Następnie dochodzi do pobudzenia neutrofilii i płytek krwi, które gromadzą się i uszkadzają otaczającą tkankę. Zachęcamy do zapoznania się z webinarium dr. Hałasy na temat nadwrażliwości pokarmowej IgG-zależnej.
Typ IV – reakcja komórkowa, reakcja opóźniona
W ten typ reakcji zaangażowane są limfocyty T, monocyty i makrofagi. Do uczulenia najczęściej dochodzi w wyniku długotrwałego kontaktu z antygenem (np. przewlekłe przyjmowanie leków, noszenie biżuterii z niklem). Tutaj pobudzane są limfocyty typu Th, które wydzielają tzw. cytokiny aktywujące limfocyty cytotoksyczne odpowiedzialne za niszczenie tkanek mających kontakt z danym alergenem. Przykładem chorób związanych z nadwrażliwością typu IV są: alergie na leki, alergie na metale, niewielki odsetek alergii pokarmowych.
Obecnie, zgodnie z zaleceniami EAACI (European Academy of Allergology and Clinical Immunology) z 2001 roku, reakcje alergiczne dzielimy już na 2 główne kategorie:
IgE-zależne (I typ nadwrażliwości)
IgE-niezależne (II-IV typ nadwrażliwości i inne)
Przebieg obu rodzajów alergii może być bardzo podobny, jednak w przypadku alergii niezależnych od IgE objawy zazwyczaj są mocno opóźnione (do 3 dni po kontakcie z alergenem, a nawet dłużej). Reakcje niezależne od IgE mają zazwyczaj mniej intensywny przebieg niż te zależne od IgE i w zdecydowanej większości nie wiążą się z ryzykiem wstrząsu anafilaktycznego, nie dochodzi również do produkcji przeciwciał IgE. Z tego powodu badania przeciwciał IgE (w tym także diagnostyka molekularna alergii) nie wykażą alergii, które od IgE nie zależą. W ich przypadku jedyną opcją diagnostyczną jest dokładna obserwacja objawów, czasowa eliminacja produktów podejrzanych o powodowanie reakcji oraz skrupulatne prowadzenie dzienniczka żywieniowego. Ewentualnie w przypadku alergii kontaktowych możemy zapytać alergologa o skórne testy płatkowe.
Alergia IgE zależna bywa także mylona z nietolerancjami i nadwrażliwościami pokarmowymi – m.in. z nietolerancją histaminy czy nadwrażliwością na salicylany. W przypadku tych zaburzeń badanie IgE także nie wykaże żadnych nieprawidłowości, mimo występowania objawów po spożyciu określonych pokarmów. Więcej na ten temat przeczytaj TUTAJ.
Alergia bez objawów?
Warto podkreślić, że o alergii mówimy wtedy i tylko wtedy, gdy występują objawy. Zdarza się, że IgE swoiste dla konkretnego alergenu pokarmowego jest podwyższone, jednak po jego spożyciu nie występują objawy. W takiej sytuacji mamy do czynienia z tzw. uczuleniem, ale nie alergią. Oznacza to, że przeciwciała IgE zostały wytworzone, jednak nie rozwinęła się pełna alergia. W takim przypadku eliminacja danego alergenu jest nie tylko niepotrzebna, ale wręcz może wyrządzić więcej szkód niż pożytku. Jeśli wyeliminujemy dany pokarm, układ odpornościowy może „zapomnieć”, że wytworzył na niego tolerancję. To z kolei może skutkować powstaniem pełnej alergii IgE-zależnej.
Podsumowując, podstawą diagnostyki negatywnych reakcji jest zawsze dokładna obserwacja objawów. Reakcje na pokarmy nie muszą być spowodowane alergią IgE zależną wykrywaną w testach alergicznych. Może wystąpić m.in. nadwrażliwość lub nietolerancja pokarmowa, które mogą przypominać objawy alergii. Ponadto, w przypadku alergii wziewnej, istnieje możliwość syntezy przeciwciał IgE na poziomie narządowym, które również nie są wykrywane w testach alergicznych. Ta złożoność mechanizmów powoduje, że każdy wynik testu alergologicznego powinien być zawsze interpretowany i skonsultowany z lekarzem, specjalistą, który uwzględni objawy kliniczne, historię pacjenta i inne czynniki diagnostyczne. To pozwoli na prawidłowe postawienie diagnozy i planowanie dalszego postępowania.
Wirus brodawczaka ludzkiego (HPV, Human Papilloma Virus) to główna przyczyna powstawania nowotworu szyjki macicy u kobiet. Warto jednak mieć świadomość, iż infekcje HPV występują również u mężczyzn i mogą mieć równie poważne skutki, doprowadzając do zachorowania na raka jamy ustnej lub prącia.
Wirus brodawczaka ludzkiego (HPV) to rodzina ok. 200 typów wirusów, wśród których znajdują się rodzaje wysokoonkogenne, zwiększające ryzyko zachorowania na nowotwór (szyjki macicy, prącia, sromu, odbytu, jamy ustnej) oraz nieonkogenne, powodujące powstawanie tzw. kłykcin kończystych. Te ostatnie nie są groźne w skutkach, jednak świadczą o występowaniu w organizmie infekcji wirusem brodawczaka ludzkiego (HPV).
Niniejszy artykuł koncentruje się na schorzeniach HPV-zależnych dotyczących narządów płciowych.
Jak dochodzi do zakażenia wirusem brodawczaka ludzkiego (HPV) u mężczyzn?
Zakażenie wirusem brodawczaka ludzkiego (HPV) jest najczęściej infekcją przenoszoną drogą płciową, można się zarazić podczas stosunku seksualnego (zarówno pochwowego, jak i analnego lub oralnego). Niestety równie groźny w skutkach może być kontakt bezpośredni z wydzielinami nosiciela (ślina) lub skórą (krocze, pachwiny, odbyt). Z tego powodu prezerwatywy poważnie ograniczają ryzyko zachorowania, ale nie chronią przed nim w 100%, bowiem wirus może być obecny poza obszarem ochrony.
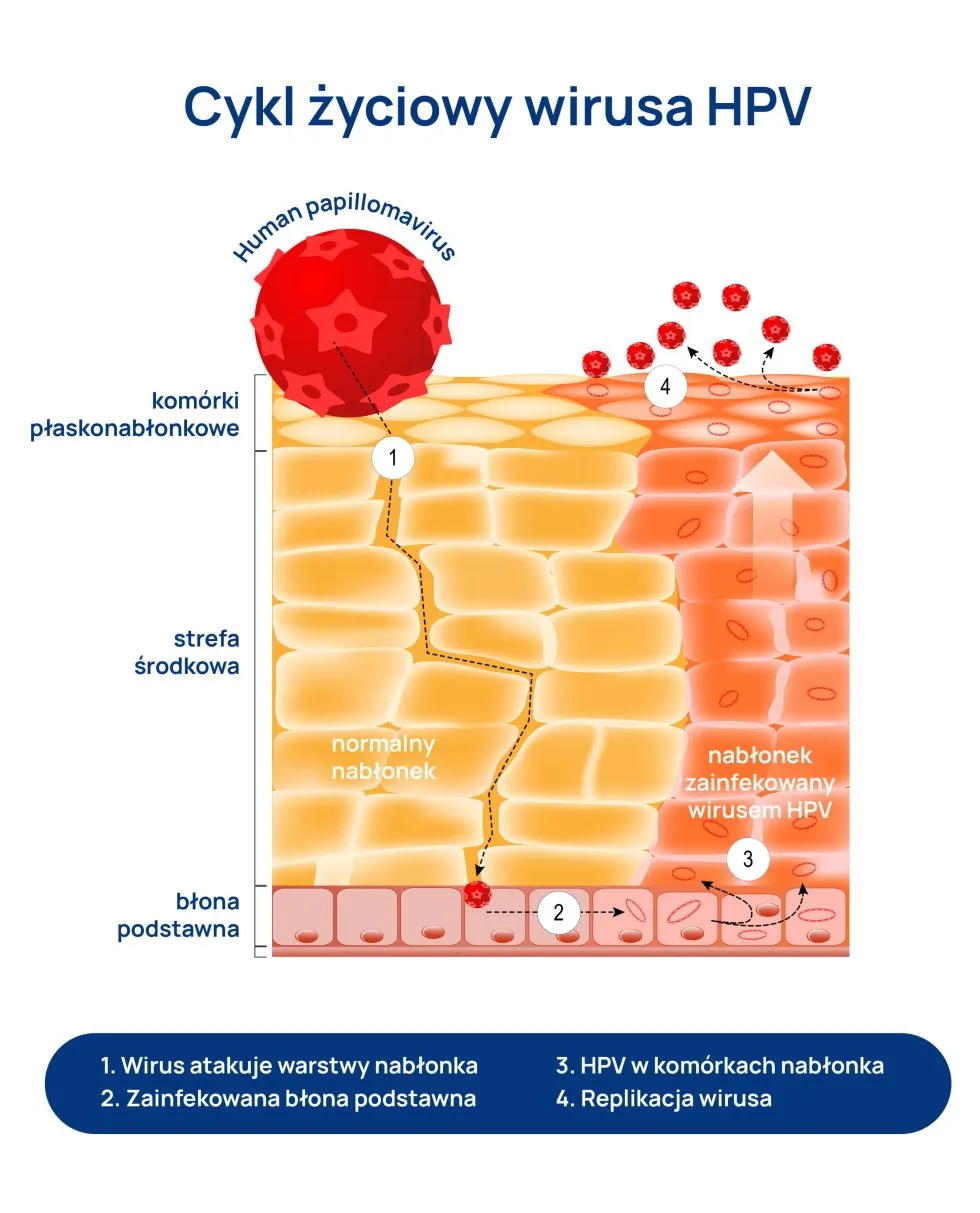
Wirus brodawczaka ludzkiego (HPV) zakaża komórki naskórka – wielowarstwowego nabłonka, stanowiącego najbardziej powierzchowną, zewnętrzną warstwę skóry. Atakuje komórki podstawne (ułożone w jednym rzędzie komórki o kształcie walca), w których ciągle zachodzą podziały komórkowe, dzięki czemu naskórek może się odbudowywać. Dlatego, jeśli namnażanie (replikacja) wirusa ma niewielkie nasilenie, infekcja sama się wygasza. Jednak w miarę dojrzewania i różnicowania się zakażonych komórek replikacja nasila się, a zainfekowane komórki ulegają transformacji i powstają charakterystyczne dla wirusa zmiany. Namnażaniu się wirusa brodawczaka ludzkiego (HPV) sprzyjają zaburzenia odporności.
Infekcja wirusem brodawczaka ludzkiego (HPV) dotyka najczęściej mężczyzn pomiędzy 18. a 39. rokiem życia, chociaż możliwa jest u każdego aktywnego seksualnie mężczyzny. Konsekwencje zakażenia mogą się również pojawiać w ciągu całego życia, np. rak prącia spotykany jest zazwyczaj u mężczyzn 60+.
Statystyki mówią, iż ok. 80% populacji (zarówno mężczyzn, jak i kobiet) było zakażone wirusem brodawczaka ludzkiego (HPV) przynajmniej raz w życiu.
Możliwe jest także zakażenie wskutek zaniedbań higienicznych, np. poprzez używanie wspólnego ręcznika.
Kłykciny kończyste to jedna z najczęstszych chorób przenoszonych drogą płciową. Przyczyną ich powstania jest infekcja nieonkogennymi typami wirusa brodawczaka ludzkiego (HPV) – 6 i 11. Oznacza to, iż kłykciny są zmianami, które nie przekształcą się w nowotwór, chociaż – jeśli występują w dużym nasileniu – mogą utrudniać funkcjonowanie i być przyczyną problemów związanych ze współżyciem (pacjenci wycofują się z aktywności seksualnej) oraz zdrowiem psychicznym (obniżenie poczucia własnej wartości, obawy związane z płodnością oraz zachorowaniem na nowotwór).
Diagnostyka kłykcin kończystych nie wymaga badań laboratoryjnych, chociaż można pobrać wymaz z żołędzi penisa, aby potwierdzić, że zakażenie wywołane jest tylko wirusami nieonkogennymi. Może zdarzyć się sytuacja, gdy źródłem infekcji jest zakażenie mieszane.
Okres inkubacji choroby – czyli czas od momentu zakażenia do pojawienia się charakterystycznych objawów klinicznych – jest zmienny, trwa od kilku do kilkunastu miesięcy. W przypadku mężczyzn kłykciny kończyste zlokalizowane są najczęściej na prąciu (na wewnętrznej blaszce napletka, na brzegu żołędzi lub w okolicach wędzidełka), rzadziej w okolicach cewki moczowej lub odbytu. Są to charakterystyczne grudkowate wykwity, często liczne. Mogą samoistnie zanikać, ale mają skłonność do nawracania. Na ogół nie powodują żadnych dolegliwości, czasem pacjent odczuwa świąd.
Leczenie kłykcin nie jest przyczynowe, nie opracowano leku zwalczającego zakażenie wirusem brodawczaka ludzkiego (HPV). U osób, które mają niewielkie zmiany, można zastosować leczenie farmakologiczne, większe i rozległe zmiany usuwa się w sposób inwazyjny (łyżeczkowanie, wycięcie, kriochirurgia, laseroterapia).
Rak prącia
Rak prącia jest rzadkim nowotworem, w Polsce występuje u ok. 200 mężczyzn rocznie. Typowy pacjent ze zmianą nowotworową tego organu ma ponad 60 lat, jednak zawsze należy pamiętać, że możliwe jest jego wystąpienie również u młodszych osób.
Jednym z podstawowych czynników ryzyka przyczyniających się do powstawania raka prącia jest brak higieny osobistej. W kulturach krajów, gdzie praktykowane jest obrzezanie chłopców, nowotwór prącia diagnozowany jest rzadziej. Tłumaczy się to faktem, iż usunięcie napletka ułatwia zachowanie higieny.
Do powstawania tego schorzenia przyczynia się również wirus brodawczaka ludzkiego (HPV) – wysokoonkogenny typ 16. Poza tym czynnikami sprzyjającymi są współistnienie zakażenia HIV, duża aktywność seksualna, stulejka i przewlekłe stany zapalne.
Pierwszym objawem nowotworu prącia, który najczęściej zauważa pacjent, jest płaska, czasem wrzodziejąca zmiana umiejscowiona na napletku lub na żołędzi. Zmiana może na początku przypominać plamkę, jedna rośnie i uwypukla się wraz z upływem czasu. Wszystkie objawy, które niepokoją, powinny być skonsultowane z lekarzem. Najczęściej nie są to zmiany w typie nowotworu, jednak warto zachować ostrożność.
W przypadku raka prącia atakowane są również węzły chłonne w pachwinach – obecność powiększonych guzków jest dla niektórych pacjentów powodem do zgłoszenia się do lekarza.
Diagnostyka nowotworu prącia zaczyna się od wywiadu oraz badania lekarskiego. O rozpoznaniu rozstrzyga biopsja. Dodatkowo lekarz zleci USG jamy brzusznej czy RTG klatki piersiowej, w celu oceny występowania przerzutów, może to być również tomografia komputerowa czy rezonans magnetyczny.
Wcześnie wykryty nowotwór prącia, w którym nie ma przerzutów, rokuje dobrze, a metody leczenia oszczędzają narząd (mogą to być krioterapia, laseroterapia, brachyterapia czy usunięcie samej żołędzi). W stanach bardziej zaawansowanych leczenie jest często radykalne i polega na częściowej lub całkowitej amputacji prącia, czyli tzw. penektomii.
Diagnostyka wirusa brodawczaka ludzkiego (HPV) u mężczyzn – jak przebiega badanie?
Wykrywanie obecności wirusa brodawczaka ludzkiego (HPV) u mężczyzn możliwe jest po pobraniu wymazu z miejsc chorobowo zmienionych na penisie lub z cewki moczowej. Jak wygląda takie pobranie? Materiał pobiera się z żołędzi penisa, najlepiej z miejsc chorobowo zmienionych – jeśli jest to możliwe, ponieważ zakażenie u mężczyzn często przebiega bezobjawowo. Próbkę pobiera się specjalną wymazówką, poprzez kilkukrotne pocieranie zmiany chorobowej ruchem spiralnym. Jeśli badanie wykonywane jest w celach profilaktycznych i nie ma widocznej zmiany chorobowej, materiał należy pobrać z rowka zażołędnego wokół penisa, na całej jej długości.
Wymaz można również pobrać z cewki moczowej, w tym celu używa się tzw. mini wymazówki, którą wprowadza się na ok. 1 cm do cewki moczowej, przytrzymuje 5-10 sekund, a następnie kilkakrotnie ją obraca.
W przypadku badań w kierunku wirusa brodawczaka ludzkiego (HPV) ważne jest przygotowanie pacjenta do badania.
48 godzin przed pobraniem wymazu nie należy stosować żadnych leków, zwłaszcza w postaci maści,
24 godziny przed pobraniem należy wstrzymać się od współżycia płciowego oraz ograniczyć zabiegi higieniczno-pielęgnacyjne okolic narządów płciowych.
Istnieje kilka typów testów, wykrywających różne typy wirusów brodawczaka ludzkiego (HPV) w różnych konfiguracjach.
Profilaktyka zakażeń wirusem brodawczaka ludzkiego (HPV) – szczepienia przeciwko HPV u chłopców
Wirus brodawczaka ludzkiego (HPV) to czynnik sprawczy groźnych nowotworów, zarówno u kobiet, jak i u mężczyzn. Rak szyjki macicy – dzięki programowi badań przesiewowych – może być wykryty wcześniej, natomiast pozostałe nowotwory HPV-zależne (gardło, prącie, odbyt) nie mają takich programów wczesnego wykrywania. Dlatego ważnym elementem profilaktyki na dziś jest szczepionka przeciwko HPV.
W Polsce dostępne są trzy typy szczepionek, a od 1 czerwca 2023 bezpłatnymi szczepieniami objęte są dziewczynki i chłopcy w wieku 12 i 13 lat. Poza refundacją zaszczepić się może również młodzież po 14 r.ż. i dorośli. Więcej o szczepionce przeczytasz TUTAJ.
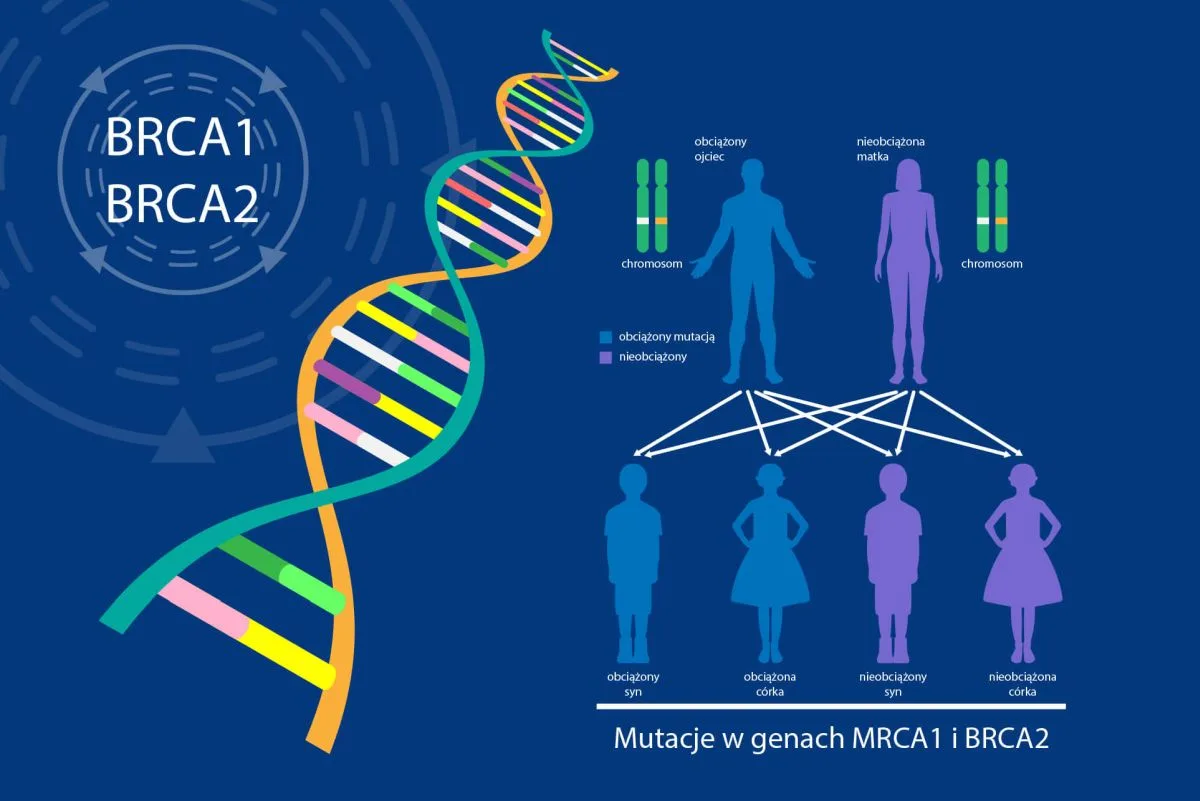
Większość przypadków raka piersi to zachorowania tak zwane przypadkowe, niezwiązane z obciążeniem dziedzicznym. Istnieje jednak niemała grupa pacjentek, jak również pacjentów (mężczyźni także chorują na raka piersi), których zachorowanie powiązane jest z nosicielstwem mutacji uszkadzającej jeden z genów chroniący przed rakiem piersi i jajnika. W ostatnich 30 latach znacznie poszerzyła się wiedza na temat przyczyn takiego stanu rzeczy. Dotychczas odkryto już kilkanaście genów, których uszkodzenia powiązane są ze zwiększonym ryzykiem zachorowania na raka piersi, jajnika i prostaty. Wśród tych kilkunastu najdokładniej przebadane i pełniące najważniejszą rolę są geny BRCA1 i BRCA2. Występujące w nich uszkodzenia powodują największy wzrost ryzyka zachorowania na raka piersi i jajnika. W przypadku raka piersi, zamiast co dziesiątej kobiety, choruje nawet do ponad dwóch trzecich nosicielek. W przypadku raka jajnika, zamiast co setnej, może zachorować nawet co druga.
Ryzyko zachorowania na raka piersi w przypadku nosicielstwa wynosi odp. 65% i 45% dla nosicielek mutacji w genie BRCA1 i BRCA2; oraz na raka jajnika odp. 39% i 11%.
U mężczyzn, którzy są nosicielami, występuje wzrost ryzyka zachorowania na raka prostaty do ponad 20%. U tych mężczyzn zdarzają się także z kilku procentową częstością zachorowania na raka gruczołu piersiowego.
Pełne sekwencjonowanie genów metodą NGS i jego zalety
Wprowadzenie technologiisekwencjonowania nowej generacji (next generation sequencing, NGS) stanowi olbrzymi postęp i przełom w możliwościach diagnostyki chorób uwarunkowanych genetycznie, w tym diagnostyki nowotworów. Należy podkreślić, że diagnostyka genetyczna genów powiązanych z ryzykiem nowotworów jest skomplikowana ze względu na ilość liter kodu genetycznego, które trzeba odczytać i przeanalizować. Gen BRCA1 składa się z około 7 000 nukleotydów, czyli literek kodu genetycznego. Gen BRCA2 posiada ich około 10 000. Wśród tych 17 000 nukleotydów wykryto już około 7 000 miejsc, w których może wystąpić uszkodzenie genetyczne, czyli mutacja.
Do niedawna, ze względu na ograniczenia technologiczne możliwe było badanie tylko wybranych pojedynczych mutacji. W wyniku badań naukowych wykryto również takie mutacje, które w polskiej populacji występują najczęściej i to one zostały włączone do pakietów badań. W większości badań wykonywanych techniką tradycyjną oferowanych na rynku jest to od kilku do kilkunastu pojedynczych mutacji. Pozostałe prawie 7000 mutacji w ogóle nie jest w tego typu testach analizowanych.
Dlatego już dziś mamy dużą grupa pacjentek, które wykonały testy starego typu i otrzymały wynik negatywny (brak nosicielstwa) mimo, że w jednej z przebadanych pozycji mogą posiadać mutację chorobotwórczą. Pozostają przez to nieświadome zagrożenia wyższego ryzyka zachorowania.
Dlaczego warto rozważyć wykonanie badań sekwencjonowaniem nowej generacji (NGS)?
W populacji polskiej za najczęstszą przyczynę występowania genetycznie uwarunkowanej predyspozycji do tych nowotworów uznaje się nosicielstwo jednej z pięciu mutacji genu BRCA1: 5382insC, 4153delA, C61G, 185delAG, 3819delGTAAA. Pozostaje jednak duża grupa pacjentów posiadających rzadkie zmiany w genie BRCA1 lub BRCA2. Zmiany te są trudne do wykrycia tradycyjną metodą sekwencjonowania Sangera, ponieważ – ze względu na wielkość obydwu genów – jest to czasochłonne i bardzo drogie. To jest miejsce dla badania technologią sekwencjonowania nowej generacji (NGS), umożliwiającą przebadanie od początku do końca wszystkich literek w każdym genie. Oznacza to, że w genach BRCA1 i BRCA2 analizowana jest każda ze znanych około 7 000 mutacji. Jest to olbrzymie zwiększenie czułości (wykrywalności) mutacji w stosunku do testów starego typu. Badanie metodą NGS pozwala na kompletną diagnostykę genetyczną genów BRCA1 i BRCA2.
Technologia sekwencjonowania nowej generacji (NGS) pozwala również na diagnostykę pozostałych członków rodziny.
Kto powinien rozważyć wykonanie badania NGS?
Wiedza o dokładnej pozycji mutacji wykrytej testem sekwencjonowania nowej generacji (NGS) u danej pacjentki pozwala w prosty sposób, testem celowanym, wykonać diagnostykę u wszystkich krewnych, którzy również mogliby dziedziczyć taką mutację. Jednocześnie pozwala potwierdzić lub wykluczyć nosicielstwo mutacji chorobotwórczej.
Dla osoby zdrowej wiedza o posiadaniu mutacji chorobotwórczej pozwala zaplanować już od młodego wieku odpowiednią profilaktykę (badania obrazowe piersi jajników – w tym USG, mammografię i wprowadzany ostatnio rezonans magnetyczny piersi) lub zdecydować się na profilaktyczną operację piersi i/ lub jajników, co pozwala zredukować ryzyko zachorowania o blisko 100%.
Kompletna genetyczna diagnostyka jest ważnym narzędziem profilaktyki zachorowania na nowotwory piersi i jajnika. Osoba, u której jednoznacznie wykluczono mutację (jest to jednoznacznie możliwe tylko w przypadku wcześniejszego wykrycia mutacji w rodzinie) powraca do grupy ryzyka populacyjnego i może stosować badania profilaktyczne zalecane dla wszystkich.
Poniższy artykuł jest zapisem webinaru, który odbył się 1.03.2023 r.
Reumatoidalne zapalenie stawów, znane również jako RZS, to przewlekła, systemowa choroba autoimmunologiczna, która atakuje przede wszystkim stawy, ale może wpływać również na wiele innych układów w ciele. Chociaż RZS jest często kojarzony z osobami starszymi, w rzeczywistości dotyka ludzi w każdym wieku, w tym tych w średnim wieku, aktywnych zawodowo.
RZS jest najczęstszą chorobą reumatyczną, często mylnie określaną jako reumatyzm. W skali globalnej szacuje się, że RZS dotyka około 1% populacji. W Polsce także mamy do czynienia z podobnym odsetkiem, co przekłada się na kilkaset tysięcy osób.
Jednym z często powtarzanych mitów na temat RZS jest przekonanie, że choroba ta obejmuje tylko stawy. W rzeczywistości RZS jest chorobą systemową, która może wpływać na wiele różnych układów w ciele, nie tylko na stawy.
Czy RZS to choroba osób starszych?
Innym często powtarzanym mitem jest wspomniane wcześniej przekonanie, że RZS jest chorobą starszych osób. Chociaż ryzyko wystąpienia RZS rzeczywiście zwiększa się z wiekiem, wiele osób rozwija chorobę w średnim wieku. Dlatego tak ważne jest szerzenie wiedzy na temat tej choroby i jej skutecznego leczenia, by pozwolić na utrzymanie jakości życia pacjentów na wysokim poziomie.
Ciąża a RZS
Częstym pytaniem dotyczącym RZS jest kwestia wpływu choroby na ciążę. W większości przypadków ciąża może przynieść ulgę w objawach RZS, choć choroba często powraca po porodzie. Trzeba jednak podkreślić, że RZS nie wpływa negatywnie na przebieg ciąży ani na zdrowie płodu. Zawsze jednak kobieta z RZS planująca ciążę powinna skonsultować swój stan zdrowia z lekarzem.
Objawy RZS
Reumatoidalne zapalenie stawów to przewlekła choroba, której cechą charakterystyczną jest zapalenie stawów. Jako choroba systemowa może wpływać na wiele różnych części ciała, nie tylko na stawy. Może dotykać m.in. narządu wzroku, układu sercowo-naczyniowego, układu oddechowego, czy układu nerwowego. Najczęstsze objawy RZS to:
Ból i obrzęk stawów, zwłaszcza stawów dłoni i stóp, stawów śródręczno-paliczkowych i międzypaliczkowych bliższych oraz śródstopno-paliczkowych. Wiele osób zauważa symetryczne zajęcie stawów, co oznacza, że jeśli jeden staw jest dotknięty, staw po drugiej stronie ciała również. Nie jest to jednak regułą.
Długa sztywność poranna – osoby z RZS często doświadczają sztywności stawów, zwłaszcza rano po przebudzeniu lub po okresie bezruchu. Taka sztywność może trwać godzinę lub dłużej.
Utrata funkcji stawów – w miarę postępu choroby, stawy mogą stać się mniej elastyczne i mieć ograniczoną ruchomość. Może to utrudniać codzienne czynności, takie jak czesanie włosów, mycie zębów czy przygotowanie posiłków.
Ogólne zmęczenie i brak energii
Niewielka gorączka
Utrata masy ciała
Nocne poty
Przewlekłe zmęczenie
Problemy skórne, z sercem, płucami, oczami i naczyniami krwionośnymi
Objawy i powikłania pozastawowe RZS
Reumatoidalne zapalenie stawów (RZS) może wpływać na wiele układów w organizmie, w tym na układ oddechowy, krążenia, nerki czy układ okulistyczny. Przykładowo, RZS może prowadzić do zapalenia opłucnej, choroby śródmiąższowej płuc, guzków reumatycznych w sercu, kłębuszkowego zapalenia nerek czy zapalenia różnych warstw oka. Czasem RZS może wywoływać objawy, które mogą być mylące, jak kaszel, zaburzenia rytmu serca czy białkomocz. Te objawy mogą wskazywać na inne schorzenia, a ich przyczyną jest RZS. Mimo że jest to rzadkie, istnieje możliwość, że objawy pozastawowe mogą być bardziej zauważalne niż objawy stawowe, szczególnie gdy leczenie stawów jest skuteczne. Ważne jest, aby pamiętać, że leczenie RZS jest złożone i wymaga koordynacji między różnymi specjalistami.
Jak diagnozujemy RZS?
Diagnozowanie reumatoidalnego zapalenia stawów (RZS) to proces, który obejmuje kilka ważnych kroków:
Wywiad: to podstawowe narzędzie diagnostyczne w reumatologii, stanowiące około 85-90% sukcesu. Lekarz rozmawia z pacjentem, starając się zdobyć jak najwięcej informacji. Ważne są pytania o objawy, takie jak: które stawy są zajęte, czy występuje obrzęk, czy występuje sztywność poranna, która może wskazywać na chorobę zapalną. Co więcej, pytania dotyczące historii medycznej pacjenta i jego rodziny są istotne, ponieważ niektóre choroby mogą prowadzić do dolegliwości stawowych, a genetyka może wpływać na predyspozycje do RZS.
Badanie fizykalne: w kolejnym kroku lekarz dokonuje badania przedmiotowego, które obejmuje ocenę stawów pacjenta. Sprawdza ruchomość stawów, czy występuje obrzęk i czy stawy są bolesne. Obrzęk może być delikatny, ale wyczuwalny pod palcami, co wskazuje na zapalenie.
Wywiad rodzinny: pytania dotyczące chorób reumatycznych w rodzinie pacjenta są również istotne. Jeśli bliscy krewni (rodzice, dzieci, rodzeństwo) cierpieli na RZS, zwiększa to ryzyko wystąpienia tej choroby u pacjenta.
Trzeba jednak pamiętać, że brak historii choroby w rodzinie nie wyklucza możliwości zachorowania na RZS. Podobnie, obecność symptomów, takich jak obrzęk stawów czy sztywność poranna, nie oznacza automatycznego diagnozowania RZS. Dlatego diagnostyka tej choroby zawsze wymaga kompleksowego podejścia i analizy wszystkich dostępnych danych.
Diagnostyka laboratoryjna w wykrywaniu RZS
Reumatoidalne zapalenie stawów (RZS) to przewlekła choroba autoimmunologiczna, która prowadzi do zapalenia i zniszczenia stawów. Diagnostyka laboratoryjna odgrywa kluczową rolę w jej wykrywaniu, zapewniając ścisłe i szybkie wykrywanie oraz umożliwiając monitorowanie postępów choroby. Podstawowe badania laboratoryjne, które są częścią diagnostyki RZS, obejmują badania krwi na obecność specyficznych przeciwciał: reumatoidalnego czynnika RF oraz przeciwciał przeciwko cyklicznemu peptydowi cytrulinowanemu (anty-CCP).
Choć badanie RF i anty-CCP jest często wykorzystywane w diagnostyce RZS, nie jest ono specyficzne tylko dla tego schorzenia. Obecność tych przeciwciał może być wykryta również w przypadku innych chorób autoimmunologicznych. Dlatego, aby zwiększyć precyzję diagnostyczną, lekarze często korzystają z dodatkowych badań, takich jak pomiar stężenia białka C-reaktywnego (CRP) i szybkości sedymentacji erytrocytów (OB), które wskazują na ogólny stan zapalny organizmu.
Rozpoznanie RZS jest procesem stopniowym i wieloaspektowym, uwzględniającym zarówno wyniki badań laboratoryjnych, jak i obraz kliniczny pacjenta. Diagnostyka laboratoryjna, mimo że nie jest jedynym elementem w diagnostyce RZS, stanowi istotne narzędzie, pozwalające na szybkie rozpoznanie choroby, monitorowanie jej przebiegu oraz efektywność stosowanego leczenia. Szczególnie ważna jest dla osób z rodzinną historią chorób autoimmunologicznych, u których ryzyko wystąpienia RZS jest wyższe.
Leczenie reumatoidalnego zapalenia stawów
Leczenie reumatoidalnego zapalenia stawów (RZS) jest skomplikowane i wielopłaszczyznowe, zaczynając od edukacji pacjenta o chorobie, jej przebiegu i potencjalnych powikłaniach, po wsparcie psychologiczne i psychiatryczne w radzeniu sobie z przewlekłym bólem i obniżonym nastrojem, które mogą prowadzić do depresji.
Ważnym elementem terapii są leki. Lekiem pierwszego rzutu jest metotreksat, który jest skuteczny, ale efekty jego działania mogą pojawić się dopiero po 6-8 tygodniach. Inne dostępne leki to m.in. leflunomid, sulfasalazyna oraz chlorochina i hydroksychlorochina. W przypadku niskiej aktywności choroby, Amerykańskie Stowarzyszenie Reumatologiczne zaleca stosowanie hydroksychlorochiny.
Kiedy te leki nie przynoszą oczekiwanych efektów, stosuje się leki syntetyczne, celowane, które wpływają na układ zapalny. To inhibitory kinazy janusowej (np. Barycytynib, Upadacytynib i Tofacytynib) – leki innowacyjne, które zmniejszają stan zapalny.
Jeszcze inną grupą leków są leki biologiczne i biopodobne, które działają przyczynowo na cząsteczki antyTNF i TNF, czynniki zwiększające stany zapalne. Przykłady takich leków to Adalimumab, Certolizumab, Etanercept.
Oprócz powyższych, w przypadku silnego bólu, stosowane są również niesteroidowe leki przeciwzapalne oraz, na krótko, sterydy.
Inne możliwości leczenia – fizjoterapia, reumatologia, wsparcie psychologiczne
Oprócz farmakoterapii, reumatoidalne zapalenie stawów można leczyć za pomocą wielu innych metod. Fizjoterapia, która obejmuje kinezyterapię, skupia się na zwiększeniu siły mięśniowej, poprawie ruchomości stawów oraz zmniejszeniu ryzyka wystąpienia niepełnosprawności. Następnie mamy fizykoterapię, której głównym celem jest zmniejszenie dolegliwości bólowych poprzez stosowanie termoterapii, balneoterapii, laseroterapii i elektroterapii. Te techniki, stosowane indywidualnie lub łącznie, przyczyniają się do złagodzenia objawów choroby.
W sytuacjach, gdy stawy pacjenta uległy destrukcji i inne metody leczenia nie przynoszą oczekiwanych efektów, możliwe jest zastosowanie interwencji chirurgicznej w ramach reumoortopedii. Ortopedzi mogą usztywnić staw lub dokonać jego wymiany, co znacznie poprawia jakość życia pacjenta.
Ponadto, niewątpliwie ważnym aspektem procesu leczenia jest wsparcie psychologiczne. Życie z przewlekłym bólem i świadomością ciężkiej choroby autoimmunologicznej może prowadzić do obniżenia nastroju i potencjalnie do poważnych zaburzeń psychiatrycznych. Wsparcie ze strony psychologów lub psychiatrów jest niezbędne w procesie leczenia, pomagając pacjentom normalnie funkcjonować mimo choroby i zapobiegając ryzyku zgonu związanego z ciężkimi zaburzeniami psychicznymi.
Czy RZS można wyleczyć?
RZS, czyli reumatoidalne zapalenie stawów, jest chorobą przewlekłą, a więc niestety nie da się jej wyleczyć. Mit, że RZS można wyleczyć, musimy obalić. Prawdziwym celem w leczeniu RZS jest osiągnięcie remisji lub utrzymanie niskiej aktywności choroby.
Remisja to stan, w którym mimo obecności choroby, nie odczuwamy jej objawów. To trochę jak z nadciśnieniem tętniczym: mimo że mamy diagnozę, stosujemy leki, które regulują nasze ciśnienie i nie odczuwamy objawów. Tak samo jest z RZS – stosujemy leki, nie mamy objawów, ale gdybyśmy je odstawili, objawy najprawdopodobniej wróciłyby. Remisja to osiągnięcie stanu, w którym choroba jest „wyciszona”, ale nie zniknęła.
Aby osiągnąć remisję, kluczowe jest realizowanie zaleceń lekarskich. Musimy stosować odpowiednie leki, w odpowiednich dawkach i formie oraz robić to systematycznie. Ważne jest również utrzymanie dobrej relacji z lekarzem, zaufanie mu i słuchanie jego zaleceń.
RZS to nie wyrok
Mimo że diagnoza RZS może być szokiem dla pacjenta, nie jest to koniec świata. Rozwój medycyny pozwolił na skuteczne leczenie tej choroby, które może doprowadzić do remisji, pozwalając pacjentom żyć normalnym życiem. Ważne jest jednak szybkie wykrycie choroby i przestrzeganie zaleceń lekarskich, w tym stosowanie przepisanych leków zgodnie z zaleceniami.
Pojęcie „próby wątrobowe” to umowne, a właściwie historyczne określenie markerów uszkodzenia wątroby i dróg żółciowych. W aktualnych wytycznych nie używa się już tego terminu, zaleca się stosowanie terminu „badania biochemiczne wątroby” (ang. liver biochemistries) albo „badania wątroby” (ang. liver tests). Do najczęściej oznaczanych badań biochemicznych wątroby należą enzymy: ALT, AST, ALP, GGTP oraz bilirubina całkowita. W tym artykule koncentrujemy się na:
AST – aminotransferazie asparaginowej,
ALT – aminotransferazie alaninowej,
GGTP – gamma-glutamylotranspeptydazie,
ALP – fosfatazie zasadowej.
Aminotransferaza alaninowa (ALT) i aminotransferaza asparaginowa (AST) to najczęściej stosowane markery uszkodzenia komórek wątroby – hepatocytów. Są powszechnie zlecane w diagnostyce chorób wątroby, traktowane jako badanie przesiewowe, decydujące o występowaniu patologii lub jej braku – oceniają integralność miąższu wątroby. Nasilenie aktywności tych enzymów zwykle odzwierciedla stopień uszkodzenia wątroby. Zdarza się jednak, że ich poziom nie koreluje bezpośrednio ze stopniem uszkodzenia tego narządu. Dzieje się tak np. w marskości wątroby, gdy ich stężenie – po początkowym okresie wzrostu – może spadać do wartości referencyjnych.
Aminotransferaza alaninowa (ALT)
Aminotransferaza alaninowa (ALT) jest enzymem występującym głównie w cytoplazmie hepatocytów, jej okres półtrwania wynosi ok. 47 godzin. Wysoką aktywność tego enzymu obserwuje się nie tylko w chorobach wątroby, ale także w komórkach mięśni poprzecznie prążkowanych, również w sercu. Niemniej jednak podwyższone stężenie aminotransferazy alaninowej (ALT) w surowicy jest dla lekarza wskazówką, aby przyczyny tego stanu poszukiwać w pierwszym rzędzie w uszkodzonych komórkach wątroby. W chorobach tego narządu obserwowane aktywności aminotransferazy alaninowej (ALT) w surowicy są wyższe niż aktywności aminotransferazy asparaginowej (AST), co jest pochodną lokalizacji tego enzymu w komórkach wątroby. Zwiększona aktywność aminotransferazy alaninowej (ALT) jest niezależnym wskaźnikiem niealkoholowej stłuszczeniowej choroby wątroby (NAFLD – non-alkoholic fatty liver disease), przemawia też za insulinoopornością.
Aminotransferaza asparaginowa (AST)
Aminotransferaza asparaginowa (AST) występuje natomiast zarówno w mitochondriach, jak i cytoplazmie komórek wątrobowych, jej okres półtrwania wynosi 17 godzin. Poza wątrobą występuje również w komórkach mięśniowych, trzustkowych, kanalików nerkowych, erytrocytach i astrocytach – komórkach glejowych centralnego układu nerwowego. Podwyższenie stężenia aminotransferazy asparaginowej (AST) jest uważane za mniej swoiste dla chorób wątroby. Schorzeniem, w którym obserwuje się wysoką aktywność tego enzymu, jest alkoholowe uszkodzenie wątroby, gdzie aktywność aminotransferazy asparaginowej (AST) rośnie bardziej niż aminotransferazy alaninowej (ALT), stosunek AST/ALT > 1.
Do najczęstszych przyczyn wzrostu aktywności transaminaz należą czynniki:
wirusowe – np. wzrost aktywności aminotransferazy alaninowej (ALT) > 35 jest wskazaniem do dalszej diagnostyki w kierunku wirusowego zapalenia wątroby typu C,
toksyczne (np. leki – zatrucie paracetamolem),
niedokrwienne,
autoimmunologiczne,
wczesna faza ostrej niedrożności dróg żółciowych.
Niezależnie od chorób wątroby zmiany w aktywnościach transaminaz związane są z działaniem wielu czynników:
zmienność dobowa, pora dnia powoduję 45% zmienność aminotransferazy alaninowej (ALT) w ciągu dnia (największą w godzinach popołudniowych). W przypadku aminotransferazy asparaginowej (AST) między godzinami 9-21 u osób z chorobami wątroby jak i zdrowych takich zmian nie zaobserwowano.
wskaźnik masy ciała BMI ma bezpośredni związek z większymi wartościami aminotransferazy asparaginowej (AST) i aminotransferazy alaninowej (ALT) (40-50% przy wysokim BMI).
wpływ wysiłku fizycznego widoczny jest szczególnie u mężczyzn – intensywny wysiłek może powodować 3-krotny wzrost aminotransferazy asparaginowej (AST) i o 20% wyższe wartości aminotransferazy alaninowej (ALT). U kobiet wzrost wartości enzymów jest niewielki <10% i większy wpływ ma trening siłowy.
w przypadku uszkodzenia mięśni następuje znaczący wzrost aminotransferazy asparaginowej (AST) i umiarkowany wzrost aminotransferazy alaninowej (ALT). W obu przypadkach zmiany są powiązane ze wzrostem wartości kinazy kreatynowej – CK
enzymy te mogą (rzadko) tworzyć kompleksy o dużej masie molekularnej (makroenzymy) z immunoglobulinami lub innymi dużymi cząsteczkami, powodując fałszywie zawyżone wyniki pomiarów.
u pacjentów z bardzo wysokim poziomem enzymów wyczerpanie substratów może być przyczyną fałszywie zaniżonego wyniku pomiaru.
Najczęściej oznaczanymi znacznikami uszkodzenia dróg żółciowych i zastoju żółci (cholestazy) są enzymy: gamma-glutamylotranspeptydaza – GGTP i fosfataza zasadowa (alkaline phosphatase – ALP).
Gamma-glutamylotranspeptydaza (GGTP)
Gamma-glutamylotranspeptydaza (GGTP) jest enzymem błonowym, jego aktywność wykazano w komórkach dróg żółciowych, wątroby, w nerkach , trzustce, sercu, płucach, jelicie cienkim, grasicy śledzionie, mózgu i gruczołach ślinowych. Wzrost aktywności gamma-glutamylotranspeptydazy (GGTP) towarzyszy przede wszystkim wielu chorobom wątroby, dróg żółciowych (jest najbardziej użyteczna do potwierdzenia wątrobowego pochodzenia wzrostu fosfatazy zasadowej –ALP) i trzustki. Rośnie między innymi w przypadku upośledzonego transportu żółci, wątrobowego stresu oksydacyjnego oraz bezpośredniej stymulacji np. przez alkohol, leki, toksyny. Szczególnie duży wpływ na aktywność gamma-glutamylotranspeptydazy (GGTP) mają leki przeciwpadaczkowe i antydepresyjne. Statyny i doustne preparaty antykoncepcyjne mogą natomiast obniżać aktywność gamma-glutamylotranspeptydazy (GGTP) w surowicy krwi.
U osób nadużywających alkoholu utrzymywanie się wysokich aktywności gamma-glutamylotranspeptydazy (GGTP) może być świadectwem braku abstynencji, chociaż tylko 70% przypadków izolowanego podwyższonego poziomu gamma-glutamylotranspeptydazy (GGTP) jest spowodowanych nadmiernym spożyciem alkoholu, a u 30% osób nadużywających alkoholu gamma-glutamylotranspeptydaza (GGTP) jest prawidłowa (poziomy te pozostają podwyższone przez 2-3 tygodnie po zaprzestaniu intensywnego spożywania alkoholu lub po uszkodzeniu wątroby). Należy pamiętać, że gamma-glutamylotranspeptydaza (GGTP) jest parametrem, który najwolniej ulega normalizacji w przypadku regresji choroby. Izolowany wzrost aktywności tego enzymu może być również pierwszym objawem pierwotnego stwardniającego zapalenia dróg żółciowych, jednak najczęściej jest cechą insulinooporności w przebiegu zespołu metabolicznego.
Fosfataza zasadowa (ALP)
Fosfataza zasadowa (alkaline phosphatase – ALP) w diagnostyce chorób wątroby jest wykorzystywana do rozpoznawania cholestazy, czyli stanu wynikającego z upośledzenia transportu lub wytwarzania żółci. Fosfataza zasadowa (ALP) katalizuje hydrolizę estrów fosforanowych w środowisku zasadowym. Enzym ten znajduje się w błonie komórkowej hepatocytów na biegunie żółciowym (a nie w komórkach nabłonka dróg żółciowych).
Fosfataza zasadowa nie jest enzymem swoistym dla wątroby: jest także produkowana w kościach, w jelicie i w łożysku. Współwystępujący podwyższony poziom gamma-glutamylotranspeptydazy (GGTP) sugeruje pochodzenie wątrobowe tej nieprawidłowości. Częstą przyczyną podwyższonych poziomów fosfatazy zasadowej (ALP) i gamma-glutamylotranspeptydazy (GGTP) jest cholestaza i indukcja enzymów wątrobowych przez alkohol lub leki. Najczęstszym pozawątrobowym źródłem ALP są kości – izolowany podwyższony poziom fosfatazy zasadowej (ALP) jest zwykle spowodowany chorobą kości (np. chorobą Pageta u pacjentów powyżej 50. r.ż., niedoborem witaminy D, przerzutami nowotworowymi) lub nadczynnością tarczycy.
Laboratorium może określić ilościowo poziom wątrobowej i kostnej izoformy fosfatazy zasadowej (ALP), jeżeli sytuacja kliniczna jest niejasna oraz gdy całkowita wartość fosfatazy zasadowej (ALP) przekracza ponad 1,5 raza górną granicę wartości referencyjnych. Najwyższe wartości fosfatazy zasadowej (ALP) (> 1000 IU/l) występują w chorobach wątroby, w których miąższ jest zajmowany przez komórki nowotworowe, amyloid, ziarniniaki lub grzybnie. W drugiej połowie ciąży istotny wzrost fosfatazy zasadowej (ALP) i pojawienie się świądu skóry są objawami cholestazy ciężarnych.
Charakter cholestatyczny wykazuje też wiele polekowych, wirusowych i autoimmunizacyjnych chorób wątroby. Przyczyną może być upośledzenie hepatocytarnego transportu lub sekrecji kwasów żółciowych, glutationu i bilirubiny bądź uszkodzenie małych przewodów żółciowych. Najczęstszą przyczyną przewlekłej cholestazy u kobiet jest pierwotne zapalenie dróg żółciowych (dawna nazwa – pierwotna marskość żółciowa). Niską aktywności fosfatazy zasadowej (ALP) mimo obecności żółtaczki obserwuje się u chorych z ciężkim uszkodzeniem wątroby w przebiegu choroby Wilsona. Niskie wartości fosfatazy zasadowej (ALP) mogą także wystąpić u pacjentów z niedoborem witaminy B12, niedoczynnością tarczycy, niedoborami cynku lub wrodzoną hipofosfatemią.
Badania biochemiczne wątroby – o czym mogą świadczyć wyniki?
Podsumowując:
Zwiększenie aktywności AST i ALT nieproporcjonalnie duże w porównaniu z fosfatazą zasadową (ALP) – świadczy o uszkodzeniu wątrobowokomórkowym.
Zwiększenie aktywności fosfatazy zasadowej ALP) nieproporcjonalnie duże w porównaniu z AST i ALT- świadczy o uszkodzeniu cholestatycznym.
W chorobach cholestatycznych wzrost stężenia bilirubiny jest objawem świadczącym o utracie ponad połowy zdolności wydzielniczych wątroby.
Izolowana hiperbilirubinemia to zwiększenie stężenia bilirubiny z prawidłową aktywnością fosfatazy zasadowej (ALP) oraz AST/ALT.
Zwykle w większości chorób wątroby, w tym w przewlekłych wirusowych zapaleniach wątroby i niealkoholowej stłuszczeniowej choroby wątroby (NAFLD) aktywność ALT jest większa niż AST.
W około 90% przypadków alkoholowej choroby wątroby aktywność AST jest większa niż ALT, a w 70% przypadków stosunek AST:ALT wynosi >2:1.
Wartość AST może być większa niż ALT także w marskości wątroby o jakiejkolwiek etiologii, ale zwykle stosunek AST:ALT nie przekracza 2:1.
Tony Badrick and Peter Turner; Review and Recommendations for the Component Tests in the Liver Function. Pubmed 2015
Titcomb CP Jr. Liver function tests: what is the risk? J Insur Med. 2003;35(1):26-35.
Hartleb M, Simon K, Lipiński M, et al. Rekomendacje postępowania u chorych z zaburzeniami czynności wątroby i kamicą dróg żółciowych dla lekarzy POZ. Lekarz POZ. 2017;3(4):225-248.
Dembińska -Kieć A, Naskalski J, Diagnostyka laboratoryjna z elementami biochemii klinicznej.WYD.Urban & Partner, Wrocław 2010.
Robert K. Murray, Daryl K. Granner, Victor W. Rodwell. Biochemia Harpera. PZWL, Warszawa 2016.
Burke MD. Liver function: test selection and interpretation of results. Clin Lab Med 2002; 22: 377-390.
Strzeszyński Ł.: Interpretacja nieprawidłowych wyników badań biochemicznych wątroby. Omówienie wytycznych American College of Gastroenterology 2017. Med. Prakt., 2017; 9: 10–24
Rak piersi w Polsce stanowi około 22,5% wszystkich zachorowań na nowotwory i jest najczęściej występującymi nowotworem złośliwym u kobiet. W przypadkuraka jajnika jest on po raku płuca, piersi i okrężnicy czwartym nowotworem odpowiedzialnym za 4,5% wszystkich przypadków nowotworów, ale jednocześnie powodującym najwięcej zgonów z przyczyn o podłożu nowotworowym. Wpływa na to brak objawów we wczesnym stadium rozwoju oraz brak badań przesiewowych, co często skutkuje diagnozą na zaawansowanym etapie rozwoju i wiąże się z wysoką umieralnością.
Obserwuje się, że około 10% zachorowań na raka piersi i raka jajnika jest związanych z dziedziczeniem uszkodzonych genów, najczęściej są to geny BRCA1 i BRCA2. Analiza nosicielstwa zmian w genach BRCA1 i BRCA2 – mutacji występujących u chorych odgrywa istotną rolę w postępowaniu zapobiegawczym i prognostycznym. Daje informację na temat ryzyka zachorowania na nowotwór, a także pozwala na wdrożenie skutecznych metod profilaktycznych oraz umożliwia wprowadzenie do opieki nad pacjentem bardziej indywidualnego podejścia.
Geny BRCA1 i BRCA2
Na podstawie informacji zapisanej w genach BRCA1 i BRCA2 powstają białka, które uczestniczą w procesach naprawy uszkodzeń DNA. Zmiany w obrębie jednego z tych genów sprawiają, że komórki dzielą się w sposób niekontrolowany, w efekcie czego rozwija się nowotwór. U osób, u których występują nieprawidłowe zmiany, obserwuje się również podniesione ryzyko zachorowania na inne nowotwory. Wśród nich możemy wymienić raka trzustki czy raka gruczołu krokowego włączając je do grupy nowotworów BRCA-zależnych. Ryzyko zachorowania na nowotwór zależne jest od rodzaju mutacji, a także jej lokalizacji w genie. Zmiany w obrębie genów BRCA1 czy BRCA2 oprócz zwiększonego ryzyka zachorowania, wiążą się z bardziej agresywną postacią oraz występowaniem nowotworu u pacjentów w młodszym wieku.
Nowotwory BRCA-zależne w 2020 roku przyczyniły się do śmierci ponad 20 tysięcy pacjentów.
Jak wskazują dane, ryzyko zachorowania na raka piersi u nosicielki mutacji w genie BRCA1 określa się na około 50-80%, a w przypadku mutacji w genie BRCA2 to około 40-60%.Dla raka jajnika zmiana w genie BRCA1 to wzrost ryzyka zachorowania na poziomie 20-40%, a dla genu BRCA2 – 40-50%.
Kiedy należy wykonać badanie genetyczne w kierunku określenie nosicielstwa zmian w genach BRCA1 i BRCA2?
Coraz częściej w diagnostyce oraz prewencji wielu chorób wykorzystuje się badania genetyczne, w tym również w profilaktyce nowotworów. Występowanie zmian w obrębie genów jest często jednym z istotniejszych czynników, mogącym wpłynąć na rozwój procesu chorobowego, a ich jak najwcześniejsze wykrycie pozwala na wprowadzenie skutecznych metod, które mogą zapobiec rozwojowi nowotworu.
Takie badanie polega na sprawdzeniu, czy w DNA osoby badanej znajdują się te nieprawidłowości, które powiązane są ze wzrostem ryzyka zachorowania na dany typ nowotworu.
Badanie obecności mutacji w genach BRCA1, BRCA2 zaleca się wykonywać u:
kobiet z rozpoznanym rakiem piersi,
kobiet, u których wykryto raka jajnika,
członków rodzin, jeśli w rodzinie potwierdzona została obecność mutacji w genach BRCA,
osób z mnogimi zachorowaniami na raka piersi i/lub jajnika w rodzinie,
osób, u których wykryto raka trzustki, jeśli posiadają krewną chorą na raka jajnika lub raka piersi, rozpoznanego przed 50. rokiem życia lub dwóch krewnych z rakiem piersi, trzustki lub prostaty.
Takie badanie może ponadto wykonać każda osoba, która chciałaby sprawdzić swoją predyspozycję do zachorowania. Materiałem do badań jest krew żylna, a samo badanie nie wymaga specjalnego przygotowania, osoba badana nie musi być na czczo.
Związek pomiędzy występowaniem mutacji w genach BRCA1 i BRCA2, a zachorowaniem na nowotwory piersi czy jajnika jest znany od dawna. Na przestrzeni lat geny te przebadano pod kątem zmian oraz częstości, z jaką występują w populacji polskiej. Na tej podstawie ustalono najczęściej występujące mutacje zwane także mutacjami założycielskimi. Wszystkie tego typu mutacje mają charakter patogenny, a więc łączą się ze zwiększonym ryzykiem zachorowania na nowotwór. Badanie genetyczne w kierunku wykrycia mutacji założycielskich polega więc na analizie kilku lub kilkunastu miejsc w obrębie genów BRCA1 i/lub BRCA2, które cechują się częstym występowaniem w polskiej populacji.
Należy zwrócić uwagę, że tego typu test jest ściśle ukierunkowany i bada tylko określone zmiany. Zgodnie z Narodowym Programem Zwalczania Chorób Nowotworowych Ministerstwo Zdrowia rekomenduje, aby w przypadku testów BRCA1 zakres badania obejmował 5 mutacji patogennych: c.5266dupC; c.181T>G; c.4035delA; c.66_67delAG; c.3700_3704 del GTAAA. Analizę można rozszerzyć o analizę większej ilości mutacji, które najczęściej występują w polskiej populacji.
Należy pamiętać, że brak mutacji założycielskiej nie wyklucza możliwości istnienia innej mutacji, która występuje w pozostałej części genu, a która nie była analizowana.
W przypadku niewykrycia nosicielstwa mutacji podstawowych (mutacji założycielskich) u osoby chorej na raka piersi i/lub jajnika, która posiada pozytywny wywiad rodzinny można rozszerzyć zakres analizy i wykonać badanie genetyczne w kierunku mutacji BRCA1 oraz mutacji BRCA2 z zastosowaniem sekwencjonowania nowej generacji (NGS). Badanie to daje możliwość zdiagnozowania wszystkich występujących mutacji w analizowanym obszarze genów u danej osoby.
Co w momencie wykrycia zmian w obrębie genów BRCA?
W przypadku stwierdzenia obecności mutacji w genach BRCA osoby znajdują się w grupie tzw. wysokiego ryzyka zachorowania. Wiedza ta umożliwia szybkie wdrożenie takiego schematu postępowania (specjalna opieka lekarska czy odpowiedni program profilaktyczny), by zmniejszyć możliwości rozwoju raka albo też wykryć rozpoczynający się proces nowotworowy jak najwcześniej.
Polskie Towarzystwo Ginekologiczne opracowało dla kobiet, które są nosicielkami mutacji w genach BRCA1 i BRCA2 rekomendacje z zalecaniami profilaktycznymi. Wśród nich znalazło się przede wszystkim częstsze w porównaniu do osób bez mutacji wykonywanie badań kontrolnych, takich jak badanie mammograficzne, badanie ultrasonograficzne piersi, rezonans magnetyczny piersi, badanie ultrasonograficzne przezpochwowe, czy oznaczenie stężenia antygenu nowotworowego 125 (CA125) we krwi.
Korzyści, które płyną z wiedzy o posiadaniu niekorzystnych zmian w genach BRCA, u zdrowych osób mogą przyczynić się do bardziej efektywnego i dostosowanego do danej osoby zarządzania ryzykiem zachorowania. Mamy możliwość pełnego wykorzystania oraz dostosowania dostępnych metod i zabiegów profilaktycznych w sposób optymalny. W przypadku pacjentów, u których nowotwór się już rozwinął, wiedza ta może pomóc w podejmowaniu decyzji terapeutycznych. Znalezienie określonych mutacji daje też niejednokrotnie informację o ryzyku wystąpienia innych nowotworów. Należy pamiętać, że na raka piersi mogą chorować również mężczyźni. Stąd niezwykle istotne jest wykonywanie badań genetycznych u osób, które mają do nich wskazania, a także poszukiwanie mutacji genetycznej wśród krewnych.
Piśmiennictwo:
Raport „Prewencja i diagnostyka nowotworów BRCA-zależnych”
Raport” Diagnostyka molekularna w leczeniu nowotworów BRCA-zależnych” MODERN HEALTHCARE INSTITUTE kwiecień 2023r.
Model prewencji oraz wczesnego wykrywania wybranych, dziedzicznie uwarunkowanych nowotworów w ramach ambulatoryjnej opieki specjalistycznej
Wojciechowska, U., Didkowska, J., Michałek, I., Olasek, P., Ciuba, A. (2020). Nowotwory złośliwe w Polsce w 2018 roku. Pozyskano z: http://onkologia.org.pl/publikacje/, dostęp z 22.02.2021
Lubiński, J. (red.) (2018). Genetyka kliniczna nowotworów wskazuje na wzrost ryzyka zachorowania na dany nowotwór.
Jacek Jassem, Maciej Krzakowski Rak piersi OnkOlOgia w Praktyce klinicznej — edukacja 2018, tom 4, nr 4




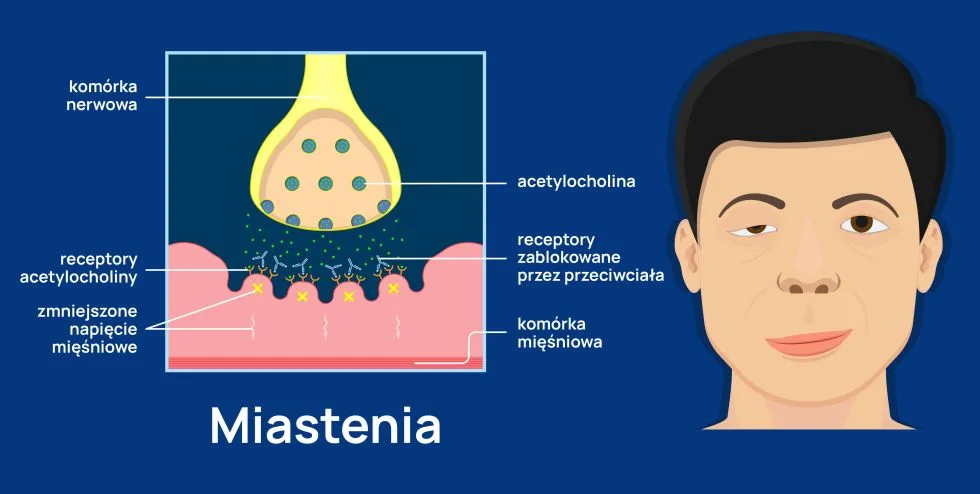




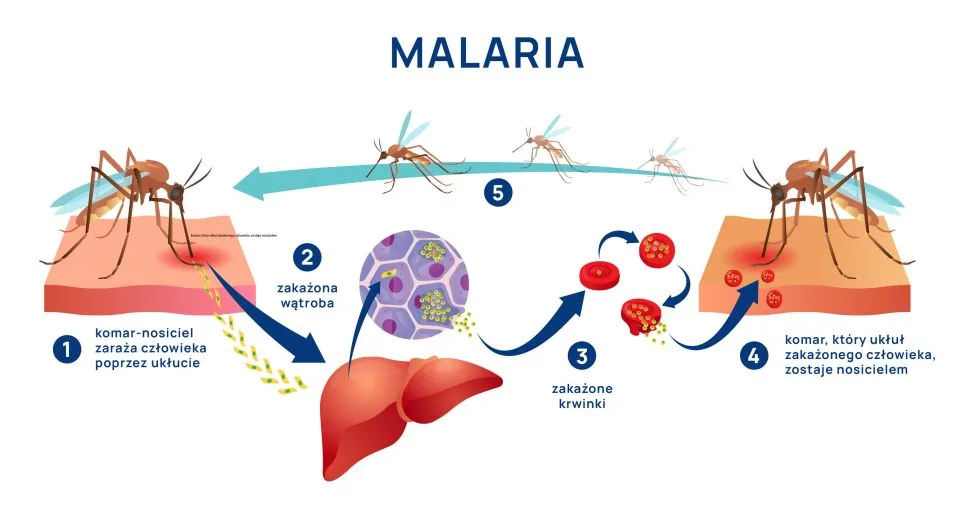
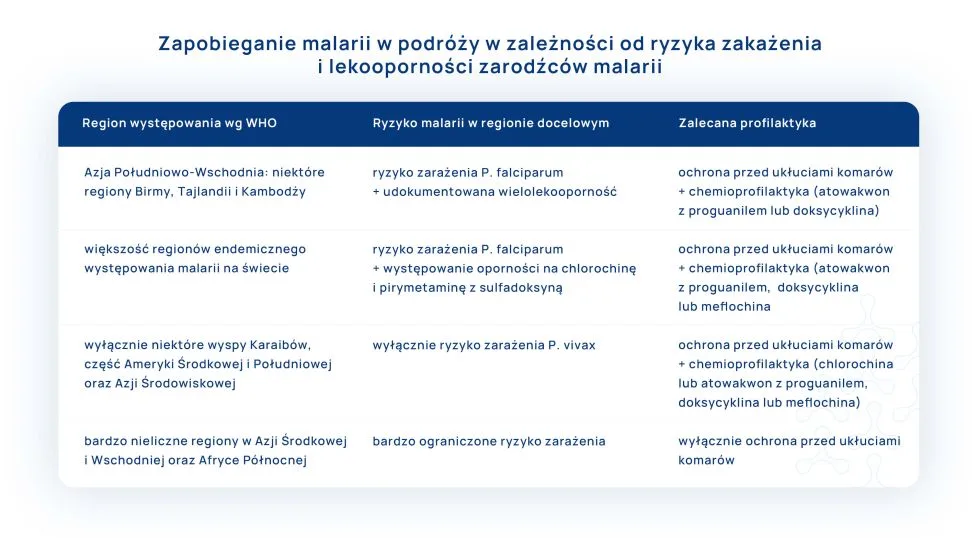



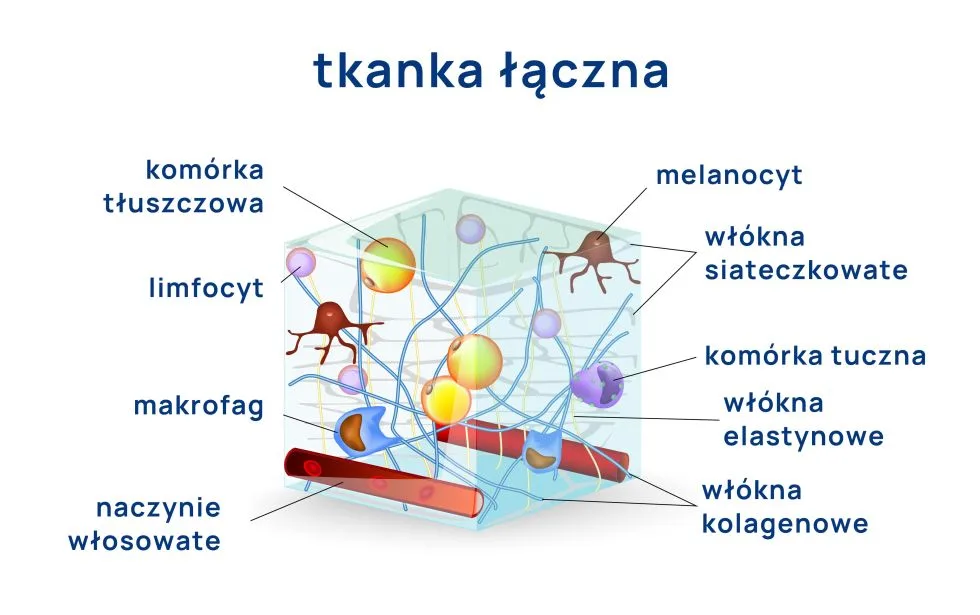


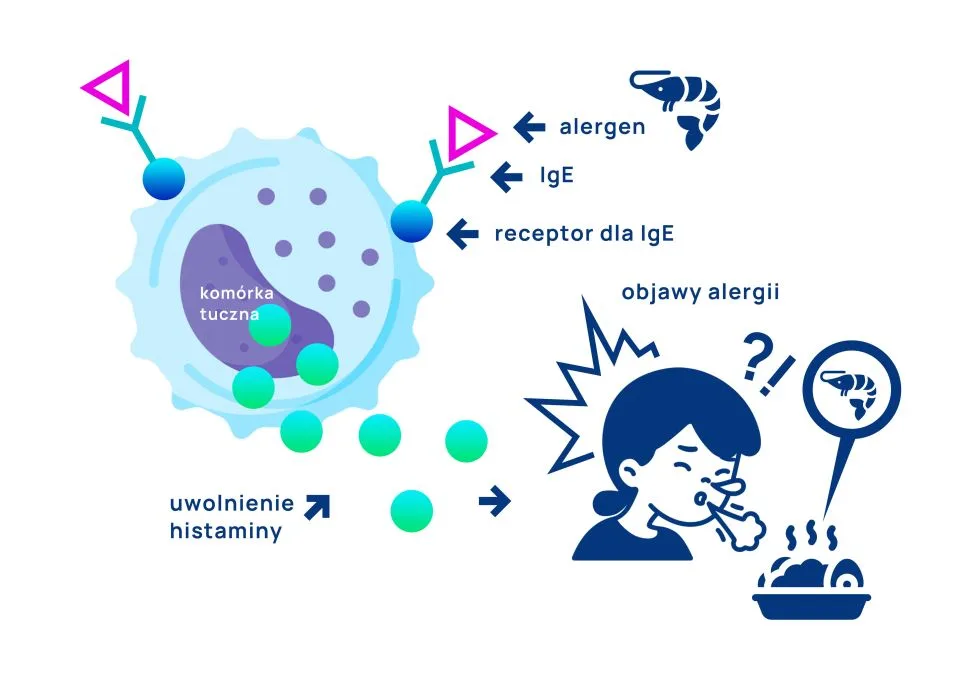


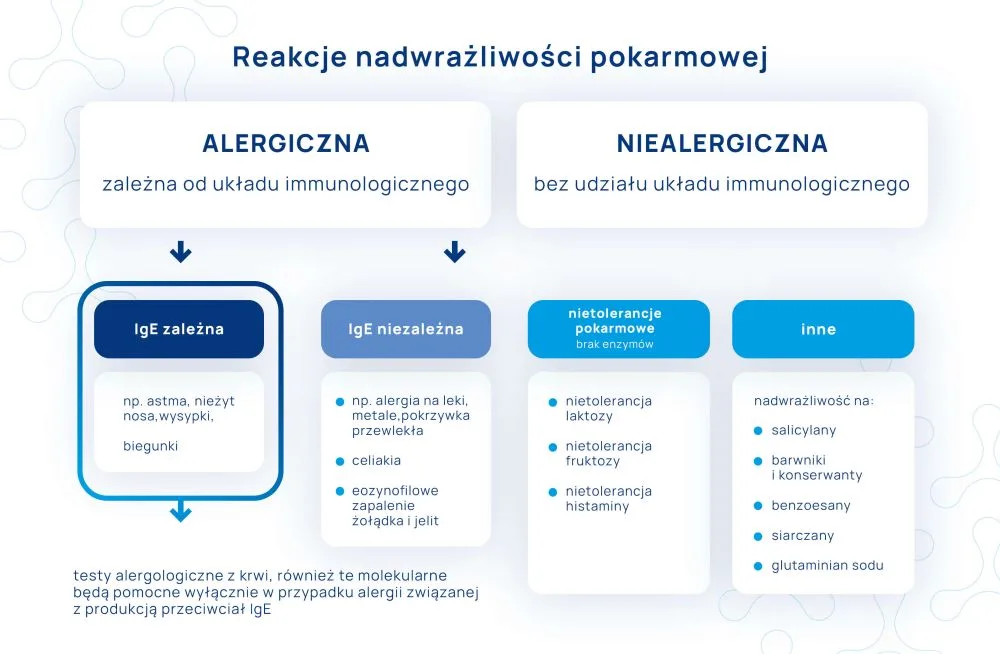
 u mężczyzn – co należy wiedzieć?")