
Układ immunologiczny (odpornościowy) człowieka jest niezwykle skomplikowanym systemem, który odgrywa rolę „strażnika”, broniąc organizm przed obcymi patogenami, takimi jak wirusy i bakterie. Jednak czasami zdarza się, że ten sam system obronny, który nas chroni, może zacząć działać na niekorzyść organizmu i taki proces nazywany jest autoagresją. Polega ona na atakowaniu własnych komórek i tkanek, co prowadzi do rozwoju chorób autoimmunizacyjnych.
Czym są choroby autoimmunizacyjne?
Choroby autoimmunizacyjne są wynikiem zaburzeń w funkcjonowaniu układu immunologicznego, które powodują, że organizm rozpoznaje własne komórki i tkanki jako obce i wrogie, co prowadzi do ich zaatakowania. Przykłady takich chorób to toczeń rumieniowaty układowy (SLE, ang. Systemic Lupus Erythematosus), reumatoidalne zapalenie stawów, cukrzyca typu 1, autoimmunologiczne zapalenie wątroby i wiele innych. Objawy tych schorzeń mogą być różnorodne i początkowo zwykle dotyczą narządów, które w danym schorzeniu są atakowane, ale z czasem proces chorobowy zaczyna również obejmować inne organy wewnętrzne.
Choć dokładne przyczyny chorób autoimmunizacyjnych nie są w pełni poznane, to istnieje wiele teorii dotyczących ich rozwoju. Jedna z nich sugeruje, że niektóre osoby mogą wykazywać pewne predyspozycje genetyczne do rozwoju takich chorób, a czynniki środowiskowe, np. infekcje czy stres, przyczyniają się do ich aktywacji.
Badania i właściwe testy diagnostyczne
Rozpoznanie chorób autoimmunizacyjnych może być trudne, ponieważ ich objawy są często niespecyficzne i mogą przypominać inne schorzenia. Do potwierdzenia diagnozy konieczne jest zazwyczaj wykonanie szeregu badań laboratoryjnych, m.in. testów w kierunku autoprzeciwciał.
Autoprzeciwciała są istotnym elementem w zrozumieniu chorób autoimmunizacyjnych. To specyficzne białka wytwarzane przez układ immunologiczny, które atakują własne komórki i tkanki organizmu, zamiast skupiać się na zwalczaniu obcych patogenów. Badania autoprzeciwciał odgrywają kluczową rolę w wykrywaniu i monitorowaniu chorób autoimmunizacyjnych, często umożliwiając wczesną diagnozę, określenie stopnia aktywności choroby czy ocenę skuteczności leczenia.
Przeciwciała przeciwjądrowe ANA (ang. antinuclear antibodies) stanowią najszerszą i często badaną grupę autoprzeciwciał. Są one skierowane przeciwko antygenom zarówno jądra komórkowego, jak i cytoplazmy. Ich ocena jest kluczowym elementem diagnostyki chorób układowych tkanki łącznej.
Oficjalne rekomendacje jednoznacznie zalecają dwuetapową diagnostykę przeciwciał przeciwjądrowych ANA. Pierwszym etapem jest test przesiewowy, który jest bardzo czuły. W tym przypadku zalecane są testy oparte o metodę immunofluorescencji pośredniej (IIFT, ang. indirect immunofluorescence technique), które są oceniane pod mikroskopem fluorescencyjnym. Doświadczony personel medyczny ocenia preparaty i w przypadku wykrycia przeciwciał przeciwjądrowych ANA rozpoznaje charakterystyczne typy świeceń, które odpowiadają poszczególnym autoprzeciwciałom. Na tym etapie można też określić poziom przeciwciał przeciwjądrowych ANA, czyli tzw. miano. Uzyskanie tej informacji jest niezwykle cenne z kilku względów:
- poziom autoprzeciwciał często jest związany z aktywnością (zaawansowaniem) choroby
- niskie miano przeciwciał przeciwjądrowych ANA (np. 1:100) może występować również u zdrowych osób

Wynik ujemny w teście IIFT jest ostateczny, podczas gdy wszelkie wyniki pozytywne wymagają potwierdzenia niezależną metodą.
Drugi etap diagnostyki, czyli etap potwierdzenia, opiera się na testach monospecyficznych, takich jak ELISA czy immunoblot, w których określa się specyficzność badanych przeciwciał. W przypadku diagnostyki przeciwciał przeciwjądrowych ANA istotne jest sprawdzenie, czy uzyskane wyniki są ze sobą spójne. Innymi słowy, przed podaniem ostatecznego wyniku, diagnosta laboratoryjny musi upewnić się, że wynik testu monospecyficznego jest zgodny z odpowiednim wzorcem fluorescencji uzyskanym na etapie badania przesiewowego.
Dla przykładu – przy podejrzeniu tocznia (SLE) zwykle wykonuje się badania w kierunku przeciwciał przeciwko:
- dwuniciowemu DNA (anty-dsDNA): przeciwciała te są obecne w kryteriach rozpoznania SLE,
- antygenowi Smitha (anty-Sm): przeciwciała te również są obecne w kryteriach rozpoznania SLE.
Powyższe przeciwciała nie są wyłącznie przeciwciałami markerowymi, ale również mają znaczenie prognostyczne i mogą być stosowane do monitorowania przebiegu SLE (ich stężenie koreluje z aktywnością choroby).
Oprócz tego w kryteria rozpoznania tocznia rumieniowatego układowego (SLE) są włączone przeciwciała antyfosfolipidowe:
- średnie lub wysokie miano przeciwciał antykardiolipinowych (IgA, IgG lub IgM),
- dodatni test na obecność przeciwciał przeciwko ß2-glikoproteinie (IgA, IgG, IgM).
Podsumowanie
Choroby autoimmunizacyjne stanowią poważne wyzwanie zarówno dla lekarzy, jak i pacjentów. Ich zrozumienie wymaga dalszych badań naukowych nad mechanizmami układu immunologicznego oraz interakcji genetycznych i środowiskowych. Obecnie kluczową rolę odgrywają laboratoria medyczne, dostarczając precyzyjnych narzędzi do diagnozy i monitorowania tych schorzeń. Dla pacjentów ważne jest zdobycie wiedzy na temat swojej choroby, regularne kontrole oraz współpraca z zespołem medycznym w celu efektywnego zarządzania objawami i poprawy jakości życia.
Piśmiennictwo


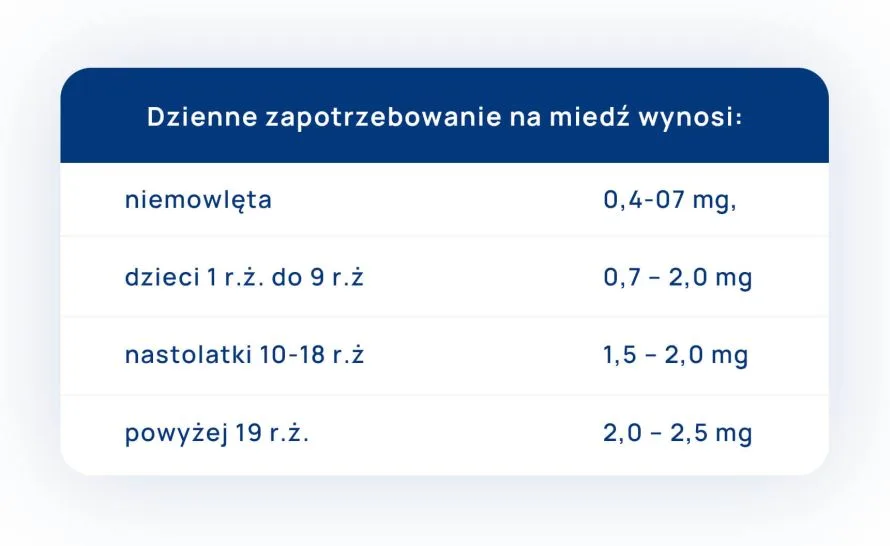
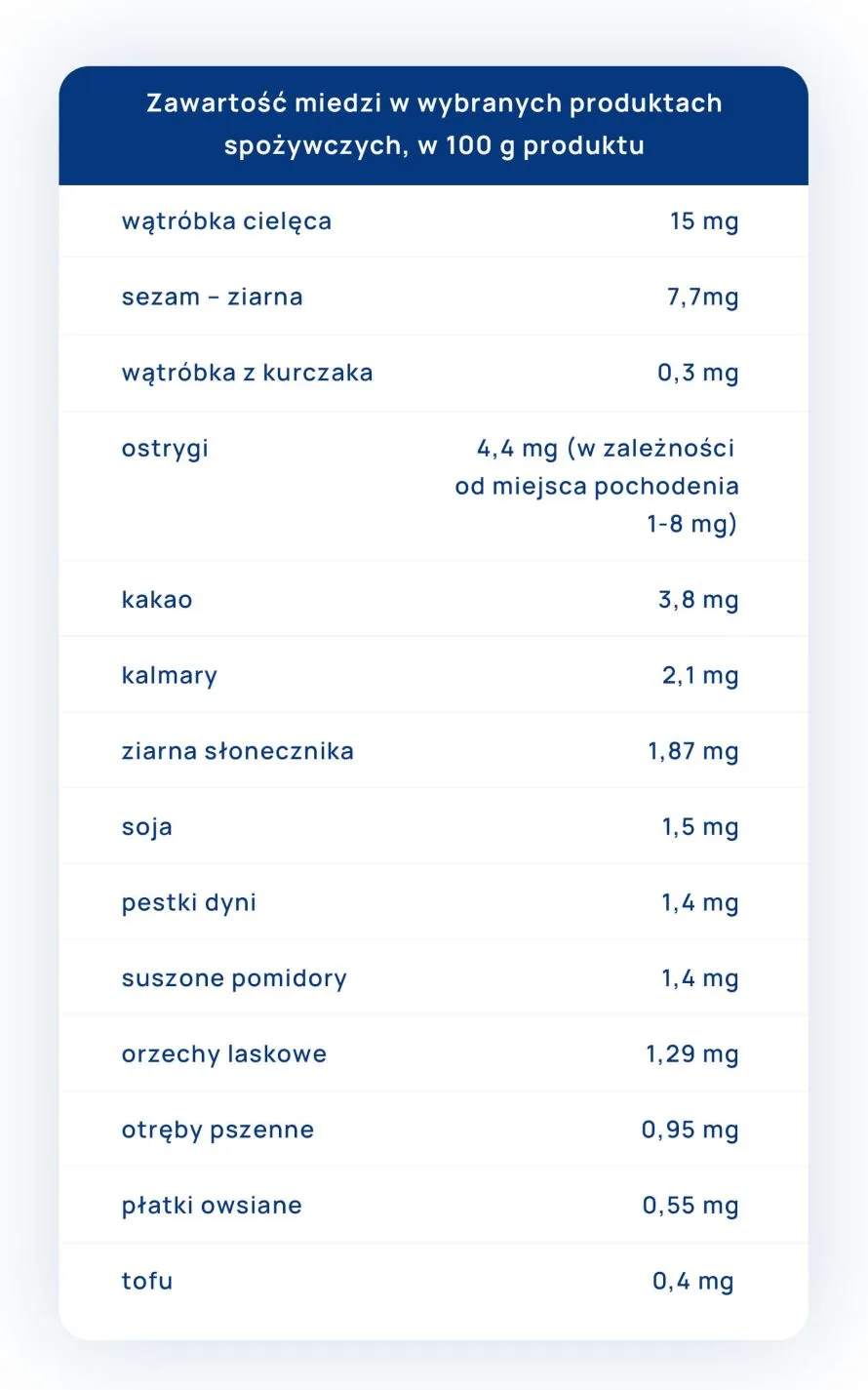
")

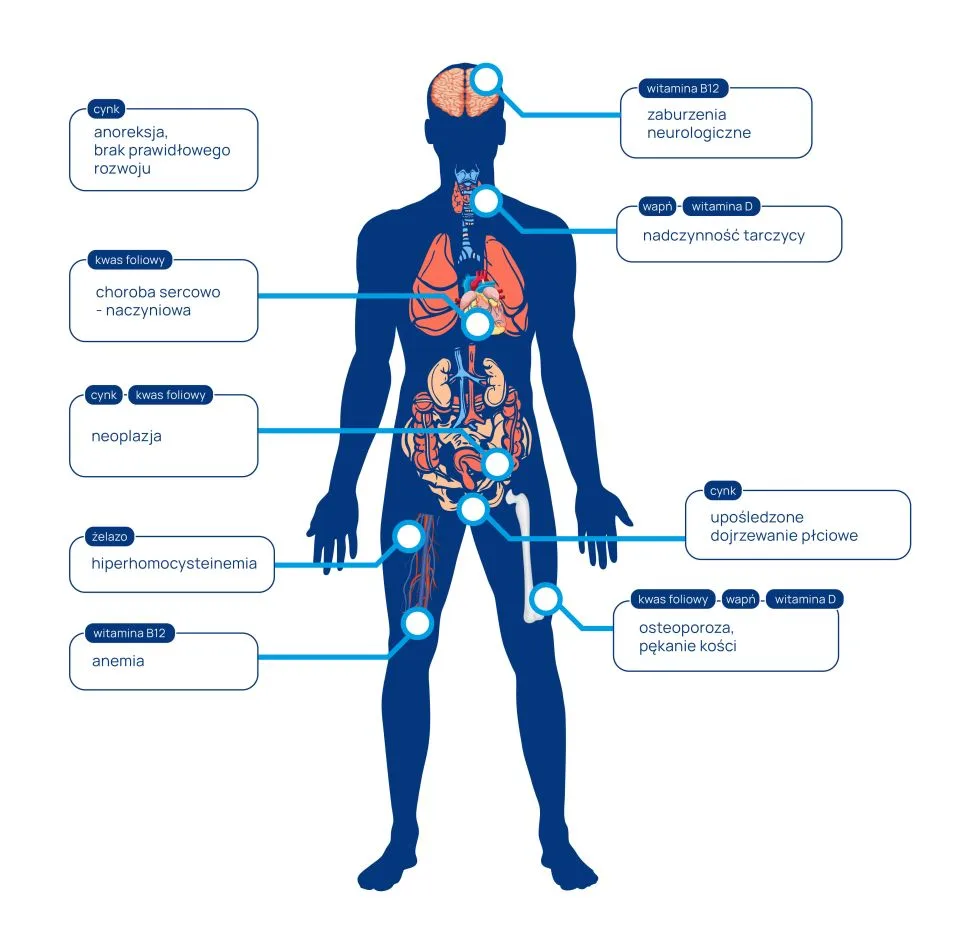



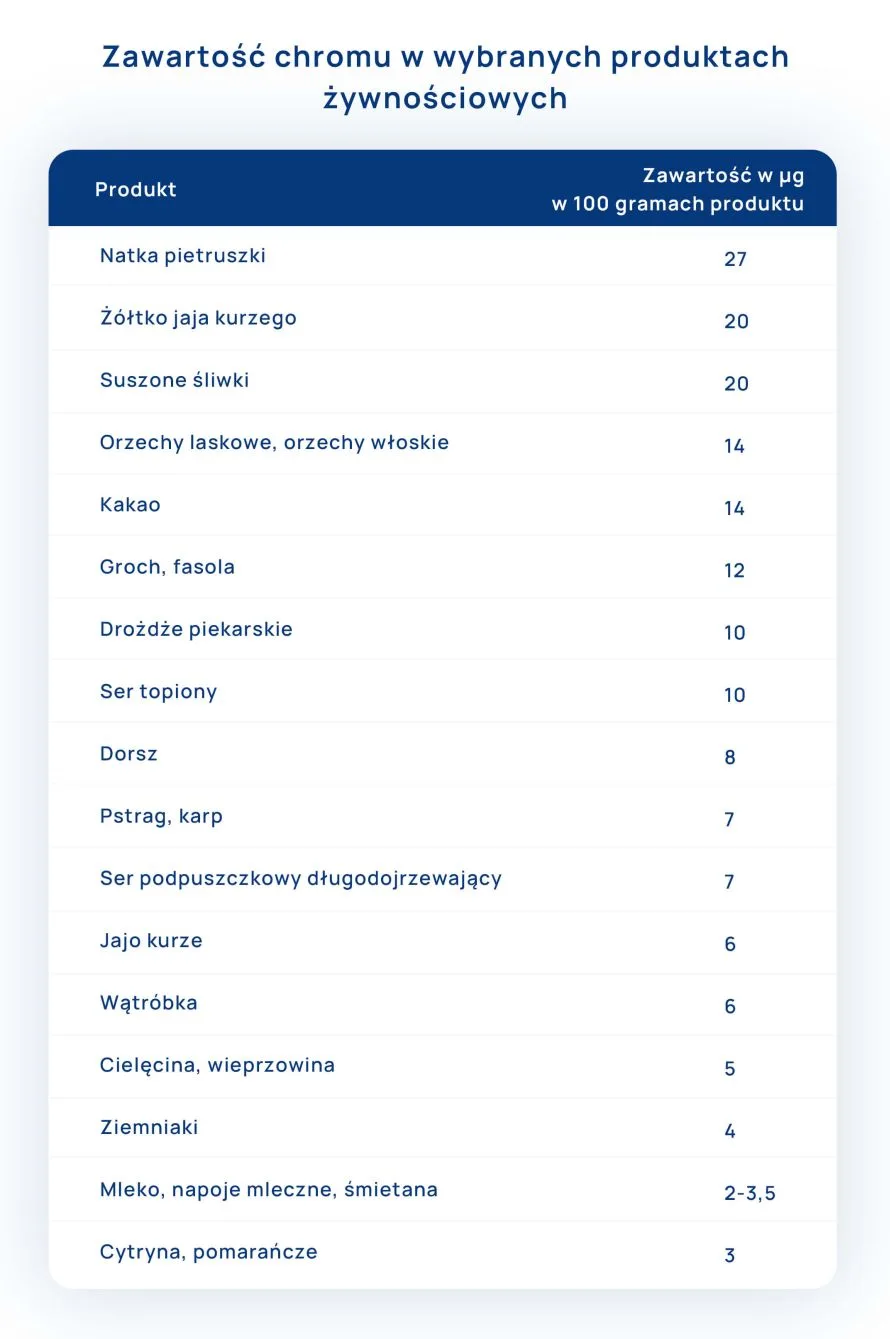










")



")


